











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Curriculum ScienTI
Curriculum ScienTI
 Permalink
Permalink
Introducción
La hemosiderosis pulmonar (HP) es una enfermedad crónica, rara, caracterizada por hemorragia alveolar y acumulación de hierro bajo forma de hemosiderina en los macrófagos alveolares, acompañada de engrosamiento de la membrana basal alveolar y fibrosis intersticial1-3.
En la mayoría de los casos se presenta en la primera década de la vida, sin predilección en cuanto a sexo1,3-6.
Puede ocurrir como enfermedad primitiva de los pulmones, denominada hemosiderosis pulmonar idiopática o primaria (HPI), o ser secundaria a vasculitis sistémicas y enfermedades cardíacas. La forma primaria es la más frecuente en pediatría, siendo su etiopatogenia poco conocida1,2,7,8.
Las manifestaciones clínicas de la HP pueden ser variables, clásicamente se presenta con la tríada de anemia, hemoptisis e infiltrados pulmonares3-6. En lactantes y preescolares suele debutar de forma aguda, acompañada de dificultad respiratoria, estertores y palidez cutáneo-mucosa debido al sangrado súbito y severo. En escolares su presentación es crónica o insidiosa, con anemia ferropénica acompañada de astenia, adinamia e hiporexia. Puede manifestarse también con hemoptisis, disnea, cor pulmonale e hipocratismo digital3,4,9,10. En oportunidades, dada su presentación clínica inespecífica, puede simular otras enfermedades, como infecciones respiratorias provocando retrasos diagnósticos1,8,6.
El lavado broncoalveolar (LBA) y/o la biopsia pulmonar confirman el diagnóstico, siendo necesaria la identificación de hemosiderófagos4,8,11.
El diagnóstico de HP idiopática o primaria se realiza por exclusión de las causas secundarias, siendo las más frecuentes en pediatría las alteraciones cardiovasculares y las vasculitis que cursan con capilaritis pulmonar (enfermedad de Wegener, síndrome de Goodpasture, etc.)9.
El abordaje terapéutico de estos pacientes requiere del seguimiento permanente de las manifestaciones clínicas y de la función pulmonar para evaluar la actividad de la enfermedad y prevenir complicaciones1.
El objetivo de este reporte es describir el abordaje diagnóstico y terapéutico de una niña portadora de SD con hemosiderosis pulmonar secundaria a poliangeitis microscópica.
Caso clínico
Dos años, sexo femenino. SD, con comunicación auriculoventricular (CIA) amplia, corregida quirúrgicamente a los 3 meses de vida. Hipotiroidismo congénito. Ectasia piélica bilateral sin dilatación uretral.
Neumonía adquirida en la comunidad con planteo de mecanismo aspirativo a los 23 días de vida. Bronquiolitis grave a los 22 meses, posteriormente sibilancias recurrentes de manejo ambulatorio, en tratamiento con fluticasona inhalada y antileucotrienos.
Un mes previo al ingreso actual, presentó episodio de fallo multiorgánico con anemia severa de perfil ferropénico de etiología no aclarada que requirió transfusión de glóbulos rojos y plasma.
Derivada al Departamento de Emergencia Pediátrica del Hospital Pediátrico del Centro Hospitalario Pereira Rossell (DEP- HP-CHPR) por fatiga de varios días de evolución. Sin síntomas respiratorios acompañantes, ni fiebre. Tránsito urinario y digestivo sin alteraciones.
Examen físico. Constantes vitales: Glasgow 15, FC 140 lpm, FR 62 rpm, Sat O2 89% ventilando espontáneamente al aire, Tax 36,7 °C, PA 90/60 mm Hg.
Fenotipo Down, regular estado general, palidez cutánea no mucosa. Bien hidratada y perfundida. Tiraje intercostal y subxifoideo con sibilancias espiratorias y subcrepitantes difusos bilaterales a la auscultación. Soplo sistólico 2/6, pulsos presentes y simétricos, extremidades cálidas. Examen abdominal y neurológico normal.
Conducta inicial: oxigenoterapia por cánula nasal de bajo flujo, broncodilatadores inhalados y corticoides vía oral. Se solicitan estudios de laboratorio y radiografía de tórax.
La radiografía de tórax mostró infiltrados algodonosos multifocales bilaterales y compromiso intersticial bilateral y difuso; el índice cardiotorácico era menor a 0,50 y la silueta cardiopericárdica no presentaba alteraciones (Figura 1). Ecocardiograma mostró CIA pequeña sin hiperflujo.

Figura 1 Radiografía de tórax, enfoque antero-pos terior. Infiltrados algodonosos multifocales bilate rales y compromiso intersticial bilateral y difuso, silueta cardiopericárdica sin alteraciones.
Estudios de laboratorio. Gasometría venosa: Ph 7,38; PCO2 37 mmHg; PO2 70,6 mmHg; HCO3 21,8 meq/L; Bass Excess -3,3; lactato 2,5 (mmol/L). Hemograma: Hb 10,4 g/dl; Hto 33,1%; HCM 23,1 pg; VCM 73,5 fl; ADE 21,6%; glóbulos blancos 10600/L, neutrófilos 6800/L (64,6%), linfocitos 1600/L (15%); plaquetas 139000/L; metabolismo del hierro: sideremia 29 ug/dl; transferrina 464 mg/dl, ferritina 93 ng/ml e índice de saturación de transferrina 5%. Reacción en cadena de polimerasa (PCR) de secreciones respiratorias: negativo para virus influenza A, B, virus respiratorio sincitial, adenovirus y SARS-CoV-2.
Dada la mejoría clínica del funcional respiratorio, se decide ingreso a sala de cuidados moderados para completar valoración.
Se solicitaron estudios complementarios: test del sudor en dos oportunidades, con resultado normal; oximetría prolongada sin y con oxígeno, con mejoría de la hipoxemia, con promedio saturación de 97%.
Tomografía axial contrastada de alta resolución (TACAR) de tórax: leve reducción de los volúmenes pulmonares a predominio de lóbulos inferiores, alteraciones en vidrio deslustrado en ambos lóbulos inferiores, língula y lóbulo superior izquierdo. Lóbulo superior izquierdo: leve engrosamiento regular de septos interlobulillares. Lóbulo superior derecho: enfisema paraseptal, posibles atelectasias laminares. Fibrobroncoscopía con LBA: vía aérea alta y bronquios hasta subsegmentarios sin lesiones. LBA: glóbulos rojos 100% conservados y escasos linfocitos. Regular cantidad de macrófagos cuyo citoplasma mostraba presencia de gránulos amarronados sugestivos de hemosiderina. No se contaba con la tinción de Wright-Giemsa para confirmarlo.
Directo normal. No se identificaron antígenos virales, bacterianos específicos e inespecíficos, ni hongos en el cultivo.
Con el planteo de HP se realizó valoración de otros parénquimas. Ecografía de abdomen y aparato urinario: hepatomegalia leve, disminución de ecogenicidad. Riñón izquierdo tercio superior e inferior con áreas hipoecogénicas mal delimitadas sin aumento de su vascularización, sugestiva de nefritis. Ectasia piélica izquierda con pelvis renal de 4 mm en sentido antero-posterior. Tomografía abdominal: hepatomegalia regular con lesión focal del segmento 5.
Funcional y enzimograma hepático: estudio de la crasis sanguínea y función renal fueron normales. Examen de orina aislado con cálculo de índice de proteinuria/creatininuria (PRU/CRU): microhematuria ++ presente en tres oportunidades, que luego se normaliza, con índice PRU/CRU de hasta 0,2 g/g.
En interconsulta con nefrólogo pediatra se descarta que la HP sea secundaria a síndrome de Goodpasture. Valorada con gastroenterólogo pediatra, dada la asociación de HP con alergias a las proteínas de la leche de vaca (APLV), se excluyen las proteínas de leche de vaca de la dieta y se indica fórmula de leche de almendras. Test de pérdida sanguínea intestinal (PSI) débilmente positivo. Segundo PSI (con exclusión dietética) negativo.
En el estudio de las causas autoinmunes se encontraron: anticuerpos de enfermedad celíaca no reactivos, dosificación de inmunoglobulina A normal. Factor reumatoideo patológico en dos oportunidades: 257 U/ml y 224 U/ml (rango: 0-20 U/ml). Complemento: C3 132 mg/dl (49-119); C4 23,4 mg/dl (17-48). Ac antinucleares (ANA) positivo (título 1/80), patrón homogéneo. Ac anticitoplasma de neutrófilo (ANCA) no se pudo realizar por presencia de Ac ANA. Ac anticardiolipinas e inhibidor lúpico negativos. Ac antimembrana basal glomerular negativa (MBG). A los dos meses se solicitan nuevamente los Ac ANCA, siendo positivos, patrón de inmunofluorescencia p-ANCA, Ac antimieloperoxidasa (MPO) positivos, Ac antiproteinasa 3 (PR3) negativo.
En conjunto con reumatólogo pediatra, dados estos hallazgos, se plantea la HP secundaria a vasculitis de pequeños vasos de tipo poliangeitis microscópica (PAM). Se indican tres bolos de metilprednisolona a 30 mg/kg/día, continuando con prednisona oral 1 mg/kg/día con descenso gradual hasta lograr la suspensión con buena tolerancia, descartando así la corticodependencia. Se realizaron siete bolos de ciclofosfamida a 500 mg/m2 como terapia inmunosupresora de inducción seguidos de ácido micofenólico como terapia de mantenimiento, habiendo cumplido 6 meses.
Presentó buena evolución clínica, sin recaídas, con mejoría del funcional respiratorio. Al mes del alta se suspende la oxigenoterapia, continúa en seguimiento con pediatra y subespecialistas.
Discusión
El caso clínico que se presenta trata de una niña pequeña con antecedente de SD que desde sus primeros meses de vida instala anemia ferropénica, acompañada de síntomas respiratorios altos y bajos recurrentes interpretados como infecciones respiratorias bajas. Uno de estos episodios se presentó con anemia ferropénica severa con repercusión hemodinámica y respiratoria, requiriendo transfusión de glóbulos rojos y plasma, no encontrándose una causa clara que lo explique. Estos hallazgos alertaron a los clínicos a analizar posibles causas de esta presentación recurrente e inespecífica. Al asociar sus antecedentes patológicos se plantearon como posibles causas de enfermedad pulmonar crónica el daño pulmonar posviral, la fibrosis quística, una probable inmunodeficiencia primaria o la HP.
Los lactantes y niños pequeños con HP no siempre se presentan con la clásica tríada de anemia, hemoptisis e infiltrado pulmonar; la hemoptisis suele faltar secundaria a la pobre capacidad tusígena9. La presentación insidiosa e inespecífica y la baja frecuencia de la enfermedad posiblemente son los factores vinculados al retraso diagnóstico, como ocurrió en este caso8.
El antecedente de ser portadora de SD tiene gran relevancia. Este síndrome favorece la aparición de enfermedades autoinmunes, como enfermedad celíaca, diabetes mellitus e hipotiroidismo. En relación con la paciente, es importante destacar que diversos estudios identifican a este síndrome como factor de riesgo para el desarrollo de la enfermedad vascular pulmonar y HP de presentación grave11-15. En los pacientes con SD, la gravedad de la HP podría deberse a la combinación de una mayor susceptibilidad a lesiones autoinmunes del capilar alveolar, sumado a alteraciones en el desarrollo de la vasculatura pulmonar vinculado a sobreexpresión de factores antiangiogénicos con mayor riesgo de hipertensión arterial pulmonar11,12,14,15. La mayor frecuencia de infecciones respiratorias bajas vinculado a niveles bajos de IgG2 sérica, conteos linfocitarios bajos y/o menor proporción de linfocitos T y NK podría vincularse a una mayor frecuencia de HP en este grupo de pacientes15.
Para confirmar el diagnóstico de HP, se necesita la demostración de sangrado intrapulmonar. La biopsia de pulmón se considera el estándar de oro para el diagnóstico; sin embargo, en presencia del hallazgo típico de macrófagos cargados de gránulos amarronados sugestivos de hemosiderina en el LBA (Figura 2), junto con un cuadro clínico compatible, como el de este caso, permitió hacer el diagnóstico de HP3. Debe considerarse que la hemosiderina en los macrófagos suele visualizarse entre 3 y 21 días luego del sangrado pulmonar, dependiendo de la entidad del sangrado16.
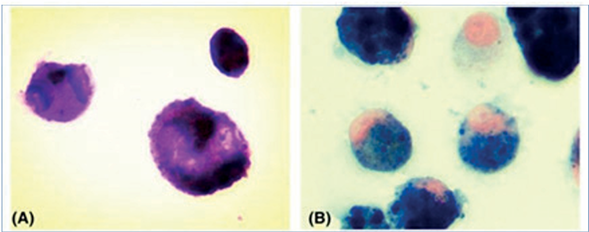
Figura 2 A. La tinción de Wright-Giemsa del líquido de lavado broncoalveolar mostró proliferación de macró fagos que contenían muchas partículas grandes en el citoplasma (x1000). B. La tinción con hierro del líquido de lavado broncoalveolar descubrió proliferación de macrófagos cargados de hierro (x1000). Imagen obtenida de Zheng L, Liu X, Zhou X, Liu K. Iron-lad en macrophages in the bronchoalveolar lavage in a welder: Pulmonary siderosis. Diagn Cytopathol 2020;48(11):1137-1140. doi: 10.1002/dc.24470
Después del diagnóstico de HP la evaluación de otros parénquimas es necesaria para determinar asociación lesional y su etiología. Por la presentación clínica, los hallazgos radiológicos y ecocardiográficos se descartó la causa cardiovascular.
El 20% de los casos de HP suelen asociar el hallazgo de hepatomegalia, como se vio en este reporte17. La presencia de nefritis y microhematuria obliga a plantear causas que asocien la afectación renal y el sangrado pulmonar. En este caso, dada la ausencia de afectación de la función renal, con microhematuria transitoria, proteinurias con índices PRU/CRU en rango normal y anticuerpos antimembrana basal glomerular negativos alejaron el planteo de síndrome de Goodpasture y por el momento se desestimó la biopsia renal.
Se detectaron marcadores de autoinmunidad como son el factor reumatoideo y Ac antinucleares positivos patrón homogéneo. La determinación de los Ac ANCA es útil para el diagnóstico de algunas vasculitis18. Los Ac ANCA son marcadores, en primer lugar, de glomerulonefritis necrotizante con semilunas aisladas, y, en segundo lugar, de enfermedad renal asociada a vasculitis sistémica de pequeños vasos. Los Ac ANCA positivos con patrón p-ANCA y la presencia de Ac anti-MPO positivos con Ac anti-PR3 negativos orientaron al planteo de la vasculitis tipo poliangeitis microscópica (PAM)19.
La presencia de microhematuria puede ser guía para la sospecha etiológica, ya que en la poliangeitis microscópica el daño renal se presenta de manera variable, con alteraciones urinarias aisladas (proteinuria o hematuria microscópica) o incluso a forma de glomerulonefritis rápidamente progresiva e insuficiencia renal20.
En niños son escasos los datos epidemiológicos acerca de las vasculitis asociadas a ANCA, se estima una incidencia de 10 a 20 casos por millón de habitantes al año, afectando mayoritariamente a individuos de sexo femenino entre los 12 a 14 años21,22.
Con respecto al tratamiento, los glucocorticoides sistémicos, como prednisona o metilprednisolona, constituyen la terapia de primera línea para el tratamiento de episodios agudos de HP. Los agentes inmunosupresores, como hidroxicloroquina, azatioprina y ciclofosfamida, se pueden asociar a la terapia con glucocorticoides orales en los casos de presentación grave5,7,11,16,23.
Respecto al tratamiento de vasculitis asociada a ANCA en población adulta y según recomendaciones extrapoladas a población pediátrica, en la fase de inducción se utilizan corticoides sistémicos asociados a ciclofosfamida o anticuerpos monoclonales anti-CD20 (rituximab), o ambos. Se ha evidenciado en ensayos clínicos randomizados que el uso de rituximab en la etapa de inducción no es inferior a ciclofosfamida endovenosa, por lo tanto, ambos fármacos son opciones a considerar en la etapa inicial de la enfermedad. En la fase de mantención o mantenimiento habitualmente se utiliza rituximab, azatioprina, metotrexato, micofenolato, entre otros. Se ha visto que con estos tratamientos los pacientes, evolucionan de forma favorable, con remisión y retiro de la terapia corticoidea sistémica, como ocurrió con esta paciente18.
El pronóstico es variable, siendo bueno en casos de presentación a forma de un único episodio con recuperación completa como en el presente caso. En aquellos con múltiples recaídas aumenta el riesgo de evolución hacia fibrosis pulmonar e insuficiencia respiratoria crónica grave3,8,5.
Es fundamental planificar un abordaje y seguimiento interdisciplinario a largo plazo que involucra en este caso a pediatra, neumólogo, reumatólogo y nefrólogo. La frecuencia y tipo de controles clínicos y paraclínicos deberán ser individualizados, no existiendo consensos internacionales. El objetivo será detectar precozmente las recaídas y tratar las complicaciones18.
Conclusiones
La HP debe ser considerada en niños que se presentan con síntomas respiratorios recurrentes y anemia ferropénica de causa poco clara, sobre todo en casos de severidad.
A pesar de que la etiología idiopática es la más frecuente, deben excluirse las causas secundarias. En este caso la hematuria microscópica alertó la posibilidad de una vasculitis, que se confirmó con la presencia de anticuerpos ANCA con el patrón específico de la poliangeitis microscópica.
Se requiere de un abordaje diagnóstico y terapéutico interdisciplinario acompañado de un seguimiento prolongado para detectar recurrencias y complicaciones.

