











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo

 Permalink
Permalink
Introducción
La hemofilia A es una coagulopatía congénita hereditaria ligada al cromosoma X, producida por el déficit del factor VIII (FVIII) de la coagulación. El tratamiento sustitutivo con concentrados de FVIII está indicado en esquemas de profilaxis para prevenir los sangrados recurrentes y en régimen episódico para la resolución exitosa de los eventos hemorrágicos en estos pacientes1.
En la actualidad la aparición de aloanticuerpos neutralizantes (inhibidores), dirigidos contra el FVIII, es la complicación más grave del tratamiento. Esta complicación es más frecuente en pacientes con hemofilia de tipo A y en las formas severas de la enfermedad (dosificación de FVIII < 1%). La prevalencia estimada de esta complicación en las personas con hemofilia A es de 20% a 30%, puede aparecer en cualquier momento de la vida, pero presenta una mayor incidencia en niños después de los primeros días de exposición (DDE) a concentrados de FVIII. Alrededor del 70% de los inhibidores se presentarán en los primeros 20 DDE y 95% dentro de los 50 DDE1-6.
El abordaje de los pacientes con hemofilia e inhibidor se debe realizar con un enfoque terapéutico bimodal dirigido a la erradicación del inhibidor mediante la inducción de tolerancia inmune (ITI) y al manejo de los eventos hemorrágicos agudos utilizando tratamiento hemostático con agentes bypaseantes. La ITI puede realizarse en cualquier etapa de la vida y consiste en la administración de altas dosis de concentrado de FVIII, en forma regular por un período de meses a años, con el fin de erradicar los inhibidores contra el FVIII. Es la mejor alternativa terapéutica para los pacientes con inhibidores de alto título al FVIII. Permite, luego de la misma, retomar el tratamiento con concentrados de FVIII para los eventos hemorrágicos, al igual que regímenes de profilaxis, y de esta manera disminuir la aparición de secuelas, mejorar la calidad de vida y disminuir la probabilidad de presentar sangrados con riesgo vital7,8.
Las probabilidades de éxito con la ITI van de 60% a 80%. El primer caso documentado de ITI fue realizado en Alemania en el año 1970, en el que un paciente con hemofilia A e inhibidor de alto título, recibió ITI y a los dos años pudo retomar la profilaxis con concentrados de FVIII9-11.
Se han descripto varios factores pronósticos que ayudan al médico clínico a predecir cómo será la evolución y el probable resultado de la ITI. Se muestran en la (Tabla 1)12-17. Estamos asistiendo a una nueva era donde aún se mantiene en consenso general que la eliminación del inhibidor es una meta importante18,19. Se ha demostrado en los estudios HAVEN 1 y 2 que el emicizumab es muy superior a un tratamiento a demanda, o a un enfoque de profilaxis con agentes bypaseantes20,21. Las preguntas a las que se enfrenta el médico hemoterapeuta clínico, cuando dispone emicizumab, más allá de si realizar o no la ITI, es cómo realizarla. Esta pregunta cuadra con el enfoque de manejo de las hemorragias. En el estudio HAVEN 2 se demostró que emicizumab reduce la probabilidad de hemorragias en niños con inhibidores en 99%22,23. Por lo tanto, si se indicara emicizumab a niños recién diagnosticados con inhibidores, esto cambiaría las consideraciones sobre el esquema de ITI más apropiado24,25. Con la disponibilidad de emicizumab existe la opción de tratar con profilaxis de FVIII o emicizumab, si bien la decisión de cambiar a emicizumab a los pacientes con ITI exitosa parece sencillo, la decisión se centra en el riesgo de recurrencia de los inhibidores. El estudio PRIORITY (NCT04621916), una vez que concluya, aportará datos al respecto.
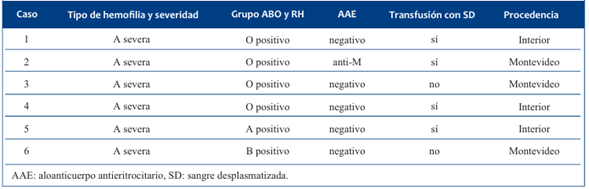
Tabla 1 Características de los pacientes (tipo y severidad de la hemofilia, antecedente transfusional y lugar de residencia).
En el Departamento de Medicina Transfusional de este prestador integral de salud se desarrolla el Centro de Referencia Nacional (CDR) para la hemofilia y otras enfermedades hemorrágicas congénitas de Uruguay. Se asiste aproximadamente al 80% de los niños con hemofilia del país, estando abocado a realizar una atención integral, acceso a diferentes especialidades y a tratamientos apropiados. El equipo multidisciplinario brinda un manejo coordinado de enfermedades crónicas y atención con expertos en hemofilia, asesoramiento de reducción de riesgos, educación continua de pacientes y familiares. A su vez, promueve el tratamiento domiciliario y la profilaxis, trabajando estrechamente con organizaciones de pacientes con hemofilia.
Iniciamos esta terapéutica con la inclusión del primer paciente pediátrico que presentaba hemofilia A e inhibidores en el año 2009 y continuamos hasta la fecha. El objetivo de este trabajo fue caracterizar a los niños con hemofilia A severa con inhibidores de alto título, tratados con ITI y describir la experiencia nacional de la ITI en el período entre 2009 y 2020.
Metodología
Se realizó un estudio descriptivo, retrospectivo, en el que se incluyeron a todos los pacientes menores de 18 años con hemofilia A severa (dosificación de FVIII < 1%) y con inhibidores de FVIII de alto título (≥ a 5 UB), a quienes se les realizó el tratamiento de ITI y seguimiento completo, en el período 2009 a 2020, en el Departamento de Medicina Transfusional del Centro Hospitalario Pereira Rossell.
La fuente de información fue la base de datos del Departamento de Medicina Transfusional y de la historia clínica de los pacientes.
Las variables analizadas fueron: tipo de hemofilia y severidad, grupo sanguíneo ABO, RhD, anticuerpos antieritrocitarios (AAE), transfusión con SD, procedencia, FVIII utilizado pre-ITI, DDE a FVIII, edad al diagnóstico del inhibidor, título en UB al diagnóstico, edad al inicio de la ITI, título en UB al inicio del ITI, tiempo de latencia diagnóstico inicio ITI, pico máximo en UB pre-ITI, uso de CVC, dosis de ITI UI/kg, intervalo interdosis, tiempo de negativización < 0,6 UB, tiempo de recuperación in vivo >65%, tiempo de recuperación de v1/2 > a 6 h, resultado de la ITI.
Las variables continuas se describieron mediante media, mediana y rango, y las discretas con frecuencias absolutas y relativas. Para la comparación de proporciones se utilizó el test de X2 y el test exacto de Fisher, en la comparación de variables se consideró significativo p<0,05.
Los regímenes de ITI fueron intensivos de altas dosis diarias o de dosis intermedias a días alternos, de forma individualizada. Los concentrados de FVIII utilizados en este tratamiento fue con FVIII derivados plasmáticos (OCTANATE). Se instalaron dispositivos de catéter venoso central (port a cath) para administrar el concentrado de FVIII derivado plasmático (dp), para garantizar y poder mantener el protocolo terapéutico sin dificultades vinculadas a la administración.
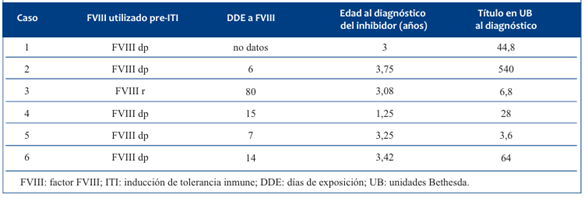
Se realizó control clínico y de laboratorio periódico, con titulación del inhibidor cada 4-8 semanas hasta que el valor fue < 0,5 UB. Una vez lograda la negativización del inhibidor se realizó la recuperación in vivo del FVIII y la farmacocinética (PK) a los 30 m, 60 m, 4 h y repitiéndolo cada cuatro semanas. Un segundo objetivo fue alcanzar una recuperación del FVIII en plasma a la hora de infusión > 65%. La definición de los resultados obtenidos según los objetivos alcanzados se muestra en la (Tabla 2)26-27.
El tratamiento de los eventos hemorrágicos antes y durante la ITI, mientras presentaban títulos de inhibidor ≥ a 5 UB, fue realizada con agentes bypaseantes (FVII recombinante activado (rFVIIa) de 90-180 µgr/kg/dosis o concentrado de complejo protrombínico activado (CCPa) de 50-100 UI/kg/dosis). La intensidad y frecuencia del tratamiento se realizó en forma individualizada, según la respuesta clínica. En los casos en que no hubo respuesta hemostática adecuada se realizó switch al otro agente bypaseante.
Una vez que los pacientes disminuyeron el título del inhibidor a < de 5 UB, se indicó para los eventos hemorrágicos concentrado de FVIII en dosis de 20 UI/kg por UB que presentaba, más el porcentaje estimado a elevar según la severidad del sangrado con una frecuencia mayor (4-6 h) y ajustado según la situación clínica.
Se solicitó aval al Comité de Ética institucional. Se solicitó consentimiento informado a los padres o tutores de los pacientes. Se resguardó la confidencialidad de los datos en todas las etapas del estudio.
Resultados
En el período de estudio cumplieron los criterios de inclusión seis pacientes con hemofilia A severa e inhibidores de alto título en el régimen de ITI, tres procedentes de Montevideo.
Cuatro de grupo sanguíneo O RhD positivo, uno A RhD positivo y el otro B RhD positivo. Cuatro presentaban el antecedente personal de transfusión con sangre desplasmatizada (SD) y uno con un AAE positivo, identificado como un anti-M. (Tabla 1) (Figura 1).

La edad promedio al realizar el diagnóstico de inhibidor fue de 2,96 años (1,25-3,75), luego de 24,4 días de exposición (DDE) (6-80) a concentrados de FVIII. En cinco pacientes correspondieron a exposición de concentrados de FVIII dp y uno a concentrado de FVIII recombinante (r) de segunda generación. En un paciente no se obtuvo el dato de DDE al concentrado de FVIII. El título en UB al diagnóstico en promedio fue de 114,5 (3,6-540). (Tabla 2) (Figura 2).

La edad promedio de inicio de la ITI fue de 3,76 años (1,66-5,08) y el tiempo de latencia del diagnóstico de inhibidor y el inicio de la ITI fue de 10,33 meses (4,0-20). El pico máximo del título pre-ITI fue en promedio de 114,7 UB (5-540). Tres pacientes necesitaron la colocación de catéter venoso central (port a cath), y en el resto se utilizó solo acceso venoso periférico.
Cuatro pacientes iniciaron el régimen de ITI con títulos de inhibidor menores a 10 UB y en los dos restantes se decidió iniciar la ITI con títulos mayores, dada la persistencia de títulos elevados y mantenidos. (Tabla 3).
Tabla 3 Edad al inicio de la ITI, características del inhibidor y necesidad de catéter venoso central.
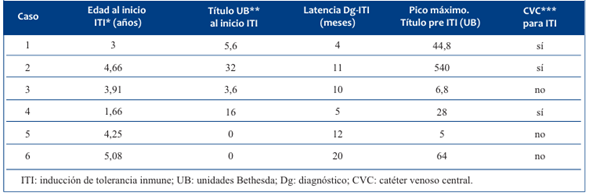
La media de dosis utilizada fue de 94,85 UI/kg (42,1-143 UI/kg), en régimen diario en dos y trisemanalmente en cuatro pacientes.
Recuperaron la vida media del FVIII en 13,6 meses (2-24) cinco pacientes. En el restante no se obtuvo una respuesta completa a la ITI, por lo que el éxito global de la ITI fue en el 83,3% de los casos.
La media para la negativización del inhibidor fue 8,2 meses (0-26). Lograron una recuperación in vivo >65 % en una media de 15,7 meses (2-33) y cinco pacientes alcanzaron una vida media >6 h, en una media 13,6 meses (2-24).
Estos objetivos alcanzados permitieron suspender el uso de agentes bypaseantes (rFVIIa, CCPa) con criterio hemostático y utilizar concentrados de FVIII en los episodios hemorrágicos (Tabla 4).
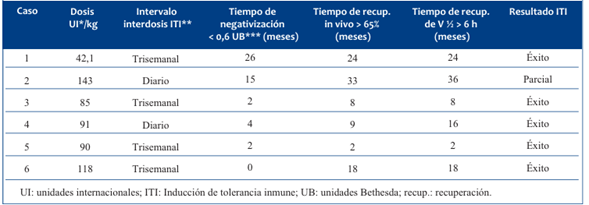
De los seis niños que lograron la inmunotolerancia, uno presentó un título histórico pre y durante la ITI > 200 UB (1.382 UB) y fue el paciente que no logró alcanzar una vida media >6 h en forma mantenida, la recuperación in vivo >65 % la alcanzó a los 33 meses, obteniendo una ITI con éxito parcial, pero logrando con la profilaxis con FVIII a días alternos una hemostasis exitosa. Un resultado extra obtenido en este paciente es que fue uno de los cuatro en recibir transfusión con sangre desplasmatizada y el único que desarrolló un AAE anti-M, lo que pone en evidencia que este paciente presenta un sistema inmune hiperreactivo muy proclive al desarrollo de anticuerpos.
Cuatro pacientes iniciaron la ITI con < 10 UB y todos con menos de 8 años de edad. El tiempo transcurrido entre el diagnóstico de inhibidor y el inicio de la ITI en todos fue menor a 20 meses.
En todos los episodios hemorrágicos que presentaron los pacientes durante el período de la ITI el tratamiento fue con rFVIIa o CCPa, en los casos en que la respuesta hemostática fue insuficiente se realizó el switch a otro bypaseante, logrando buena hemostasis, por lo que en ningún caso requirió tratamiento secuencial o concomitante y no existieron complicaciones ni eventos trombóticos.
En todos los niños una vez finalizada la ITI (ITI exitosa o parcial) pasaron a profilaxis con FVIII dp trisemanal. El descenso de la dosis fue gradual, hasta alcanzar las dosis próximas al régimen de profilaxis habitual (30 UI/kg dosis), controlando que el nivel valle de FVIII fuera > 1,5% a las 48 o 72 h. En el niño con éxito parcial la dosis se mantuvo por encima de 50/UI/kg en días alternos sin presentar eventos hemorrágicos. Durante el período analizado no se presentó reaparición de los inhibidores una vez finalizada la ITI.
Discusión
En los pacientes con hemofilia A e inhibidor no existen dudas de que se debe realizar la erradicación del inhibidor mediante ITI. Es la mejor alternativa terapéutica para los pacientes con inhibidores de alto título al FVIII, permitiendo una vez concluida la ITI, tratamientos sustitutivos con concentrados de FVIII para los eventos hemorrágicos, al igual que para la profilaxis. La ITI puede realizarse en cualquier etapa de la vida y consiste en la administración de altas dosis de concentrado de FVIII, en forma regular por un período de meses a años, con el fin de erradicar los inhibidores contra el FVIII10,11.
En el período de estudio no se contaba con emicizumab, molécula que está iniciando una nueva era en el tratamiento de la hemofilia A y determinará consideraciones sobre cuál será el esquema de ITI más apropiado19.
La elección del esquema de ITI más adecuado se determinó en base a la disponibilidad de concentrados de FVIII en el Departamento de Medicina Transfusional del prestador integral de salud y en el país, en contar con el consentimiento informado de los padres/tutores y en el compromiso de cumplir el régimen sin interrupciones. En los niños con pobre capital venoso y régimen más intensivo se requirió la colocación de catéter venoso central, conociendo las complicaciones que conlleva. Los pacientes con régimen más intensivo presentaron menores sangrados.
Dentro de los factores de buen pronóstico de la ITI está el inicio con <10 UB y con un pico histórico de inhibidor < 200 UI. En dos pacientes incluidos en el estudio se inició la ITI con un título > 10 UB, y uno de estos tenía un pico histórico pre y durante la ITI > 200 UB, el cual logró éxito parcial. Una vez que los pacientes comenzaron a negativizar el inhibidor y a medida que se fue mejorando el índice de recuperación in vivo, se pudo manejar la hemostasis con dosis altas de FVIII frente a los episodios hemorrágicos y en procedimientos invasivos intercurrentes. Se logró ITI exitosa en el 83,3%, valor comparable con publicaciones internacionales. El éxito lo atribuimos en parte a que no existieron períodos sin interrupciones de la ITI mayor a dos semanas, que fue realizada con concentrados de FVIII dp y no se realizó switch de concentrados de FVIII de diferentes presentaciones comerciales26,27.
En referencia a la inmunogenicidad de los concentrados de FVIIIdp y FVIIIr, el paciente que recibió FVIIIr de segunda generación desarrolló el inhibidor luego de 80 DDE y tuvo un título con un pico bajo de 6,8 UB. Durante el período de estudio fueron muy pocos los pacientes que recibían FVIIIr (< del 10% de la población total) y fue el único que desarrolló inhibidor. Hubiese sido esperable en este paciente el desarrollo del inhibidor más precoz con un título más elevado, al compararlo con los que recibieron FVIIIdp, y posiblemente una prevalencia de desarrollo de inhibidor mayor en los pacientes tratados con FVIIIr de segunda generación. El tamaño de la muestra de estudio es muy pequeño como para sacar conclusiones estadísticamente significativas al respecto.
Recientemente se ha aprobado el uso de emicizumab, que es un anticuerpo monoclonal humanizado biespecífico, para regímenes de profilaxis en hemofilia A con y sin inhibidores. Presenta muy buenos resultados, seguridad y excelente capacidad hemostática, disminuyendo la tasa anual de sangrados en 99%, a su vez, es de administración S/C semanal o quincenal, o mensual una vez realizada la dosis carga, por lo que está cambiando la perspectiva terapéutica19-27.
Consideramos que disponer de emicizumab no sustituye la indicación de realizar ITI, solo replantear cuál es el esquema de ITI más adecuado para el paciente y qué régimen de profilaxis utilizar una vez lograda la ITI exitosa, si con concentrados de FVIII o con emicizumab.
En todo paciente con inhibidor el mejor escenario es la erradicación del inhibidor mediante ITI, permitiendo que, una vez lograda la inmunotolerancia, se pueda utilizar los concentrados de FVIII con indicación sustitutiva frente a episodios hemorrágicos. Esto permite no depender de la necesidad de bypaseantes, los cuales no son tan eficientes desde el punto de vista hemostático, son costosos, poco disponibles a nivel sanitario y, por último, al lograr la negativización del inhibidor, permite eliminar una contraindicación para la indicación de terapia génica a futuro.
Conclusiones
La ITI es el tratamiento de elección para los pacientes con hemofilia A e inhibidores.
Durante el período de estudio, se observaron tasas de éxito muy altas similares a lo reportado en publicaciones internacionales. El paciente que tuvo una respuesta parcial presentaba elementos de mal pronóstico, lo que reafirma la importancia de considerar estos factores descriptos internacionalmente.
Dentro de los logros más importantes a lo largo del proceso de investigación, fue comprobar que se pudieron realizar en este centro tratamientos y seguimientos de pacientes con hemofilia tan complejos durante más de una década, obteniendo resultados terapéuticos comparables al de centros de gran porte a nivel mundial.
Consideramos que este estudio es muy importante para nuestro medio, ya que muestra la experiencia nacional de la ITI en pacientes pediátricos con hemofilia A e inhibidores en un período de 11 años. El trabajo es inédito y presenta esperables limitaciones propias de tratamientos tan específicos y complejos realizados en enfermedades raras de baja prevalencia.
Alentamos poder ampliar y continuar la investigación presentada, ya que se están incorporando nuevas moléculas para el tratamiento de la hemofilia, que están cambiando la perspectiva terapéutica y probablemente también los regímenes de la ITI.

