











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo

 Permalink
Permalink
Introducción
¿Qué se sabe?
Se sabe que las encefalopatías de este tipo tienen muchos efectos negativos sobre el neurodesarrollo, llevando a deterioro cognitivo severo, y que deberíamos sospecharlas mucho más. Esto llevaría a un tratamiento adecuado y así a un mejor pronóstico de estos pacientes.
¿Qué aporta este artículo?
Aporta el caso de un paciente cuyo interés radica en la presentación típica de la enfermedad descrita en la literatura, pero de sospecha y diagnóstico temprano, lo cual no es habitual. Y así redireccionar el tratamiento, utilizando bloqueadores de canales de sodio que conllevan a un mejor manejo de las convulsiones, y no de fenobarbital y levetiracetam, que son los anticonvulsivantes más utilizados en neonatología.
Las enfermedades relacionadas al gen KCNQ2 son un grupo de enfermedades epilépticas que se inician en el período neonatal, habitualmente dentro de las primeras cuatro semanas de vida de un niño. Dentro de este grupo de distintas patologías, el cuadro clínico puede variar. La más leve es la epilepsia neonatal familiar benigna (ENFB) y la más grave es la encefalopatía epiléptica (EE)1.
Dado que estas mutaciones son poco conocidas para tener en cuenta a la hora de diagnosticar epilepsias neonatales, presentamos un caso con alteraciones en KCNQ2 y desarrollo de convulsiones en el período neonatal temprano, con el objetivo de visibilizar el beneficio que conlleva el diagnóstico temprano de estos pacientes.
Caso clínico
Se evalúa en la emergencia pediátrica un neonato de 11 días de vida que presenta en el domicilio episodios tónico clónicos de miembro superior derecho asociados a cianosis peribucal, los cuales se pueden observar mediante video filmado por sus padres.
Al ingreso a UCIN se constata tono disminuido, y a los 10 minutos presenta nuevo episodio tónico clónico de miembro superior derecho y miembro inferior izquierdo, asociado a cianosis peribucal, se administra carga de fenobarbital y se decide su internación con diagnóstico inicial de síndrome convulsivo neonatal para estudio. No se encuentran antecedentes perinatológicos de jerarquía. Se inicia tratamiento anticonvulsivante con fenobarbital mantenimiento junto a la incorporación de piridoxina y biotina. Se realiza laboratorio de control a su ingreso, hemograma, medio interno y screening de infección dentro de parámetros normales. Se interconsulta a neurología infantil, que indica plan de estudio que contempla laboratorio inicial de urgencias neurometabólicas (glucosa, amonio, ácido láctico, ácido pirúvico, metabolitos en orina, pesquisa neonatal, aminoácidos en sangre, acilcarnitina total y libre), y mientras se esperan resultados y el paciente continúa con episodios paroxísticos (en esta oportunidad hipotonía y fijación de la mirada en varias oportunidades), se realizan nuevos estudios de neuroimagen, registro electroencefalográfico y panel genético para convulsiones neonatales tempranas. Se escala tratamiento y se suma levetiracetam y posteriormente vigabatrina.
Esquema de tratamiento: vigabatrina 50 mg/kg, fenobarbital 5 mg/kg/día, levetiracetam 50 mg/kg/día y biotina 5 mg/día.
Estudios complementarios realizados
- Laboratorio: hemograma, función renal, medio interno y hepatograma dentro de parámetros normales. Estado ácido base normal: 7,37/37/21/-3,2.
- Laboratorio neurometabólico dentro de parámetros normales: amonio 151 µ/dL, ácido láctico 1,6 mg/dL, glucemia 78 mg/dL, ácido pirúvico normal, metabolitos en orina normal, pesquisa neonatal normal, aminoácidos en sangre normal, acilcarnitina total y libre normal.
- Serologías del embarazo actualizadas: negativas.
- Pesquisa neonatal (seis determinaciones): normal.
- Pesquisa para enfermedades neurometabólicas ampliada: normal.
- Hemocultivos: negativos.
- Fondo de ojos: normal.
- Ecografía cerebral: normal.
- Ecocardiograma: normal.
- Resonancia magnética de cerebro con protocolo de epilepsia con espectroscopía: sin hallazgos patológicos de relevancia.
- Tomografía axial computada de cerebro: normal.
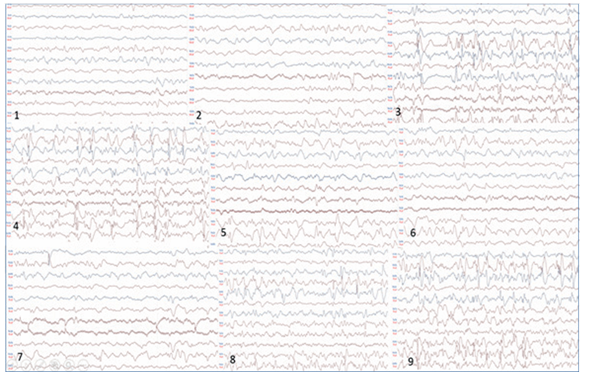
- Polisomnografía con oximetría de pulso: trazado de vigilia y sueño con abundantes espigas y poliespigas, ondas centrales izquierdas con propagación a regiones anteriores que por momentos se organizan en trenes y temporales izquierdas independientes, frecuentes poliespigas ondas fronto-centro-parietales derechas y temporales derechas independientes. Se evidenció una crisis de inicio focal electrográfica. Eficiencia de sueño disminuida. Sin alteraciones respiratorias. Sin movimientos de piernas en rango patológico (Figura 1).
- Panel genético para convulsiones neonatales tempranas: alteración para el gen KCNQ2.
Discusión
Dentro de estas enfermedades relacionadas con el gen KCNQ2 hay distintas variantes, la más leve del grupo es la ENFB, caracterizada por convulsiones en niños sanos alrededor de los 3 días de vida y que por lo general desaparecen dentro del primer al cuarto mes, la más grave es la EEP. Esta se caracteriza por epilepsia y discapacidad intelectual profunda. El tratamiento de las convulsiones es de difícil manejo, pero, por lo general, desaparecen en meses o años, también pueden volver a aparecer en la infancia. Es una patología autosómica dominante1.
Las ENBF, asociadas a mutaciones en KCNQ2, son en su mayoría breves y rara vez cursan con estatus epiléptico, no es muy habitual tampoco que desarrollen epilepsia a lo largo de la vida2.
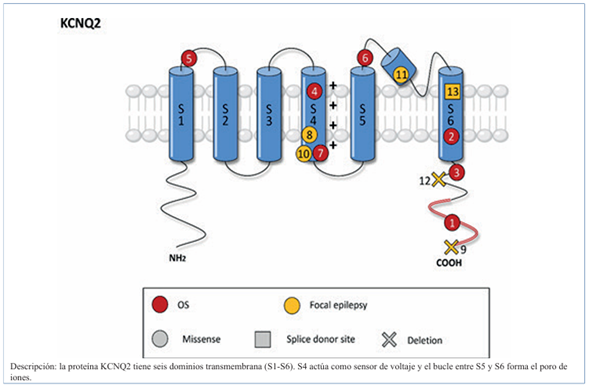
Dicho gen codifica una subunidad formadora de canales de potasio que se encuentran en las neuronas, éstas transmiten señal eléctrica (corriente M) y así las neuronas no permanecen constantemente activas. Mutaciones en el gen pueden desencadenar excesiva excitabilidad neuronal causando CNBF o EEP 7, o ambas3.
Gracias a la evolución de los estudios genéticos y a la medicina de precisión, el diagnóstico de estas patologías epilépticas de causa genética es posible y es beneficioso para el paciente el tratamiento temprano, dado que puede evitar el efecto que tiene sobre el neurodesarrollo4.
La encefalopatía KCNQ2 se presenta en la etapa neonatal temprana con convulsiones intratables, generalmente convulsiones tónicas con patrón electroencefalográfico de supresión en ráfaga y retraso grave en el desarrollo5-8. Las convulsiones intratables y el mal pronóstico justifican la necesidad urgente de un tratamiento eficaz.
Recientes estudios clínicos observaron la eficacia de bloqueadores de los canales de sodio (BCS) en algunos pacientes con encefalopatía KCNQ2 al reducir la hiperexcitabilidad causada por pérdida funcional del canal de potasio9,10.
Los pacientes con encefalopatía con alteración del gen KCNQ2 mostraron eficacia a los BCS en el control de las convulsiones. Sugerimos el inicio de tratamiento temprano con BCS en las convulsiones neonatales cuando se sospecha encefalopatía por KCNQ2, en lugar de fenobarbital o levetiracetam, que son los anticonvulsivantes más utilizados en convulsiones neonatales11 (Figura 2).
Conclusiones
Conocer los genes causantes de las distintas formas de epilepsia ampliará el conocimiento acerca de las vías moleculares involucradas en el origen de dichas patologías y además podría tener una gran repercusión en el diagnóstico, pronóstico y tratamiento de las crisis11-13.
La medicina de precisión es muy importante para diagnosticar y tratar a cada paciente como único, de manera individual, es por esto que hacemos hincapié en estudios moleculares, como en el caso citado, y así poder brindar una atención médica más precisa y efectiva.

