











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo

 Permalink
Permalink
Introducción
Una de las causas de falla cardíaca y síncope es la miocardiopatía hipertrófica (MCH), una enfermedad cada vez más diagnosticada debido al avance en las técnicas de diagnóstico y la sospecha clínica. Se presenta un caso de variante genética patogénica refractaria al manejo médico convencional.
Caso clínico
Paciente masculino de 60 años con cuadro de dolor torácico y disnea (clase funcional NYHA III/IV). En 2005 se diagnosticó MCH asociada a variante MYBPC3 c.1102A>T (p. Lys368*) asociada con un codón de parada prematura con patrón de herencia autosómica dominante asociado con MCH tipo 4. Actualmente se encuentra en manejo con metoprolol succinato y desde 2007 usuario de cardiodesfibrilador para prevención primaria. Antecedentes: neuropatía de fibra pequeña en miembros inferiores y fibromialgia controladas. A la revisión por sistemas síncope a repetición, a pesar de adecuado funcionamiento del dispositivo y adherencia al tratamiento farmacológico. Examen físico: soplo sistólico grado III/VI en foco aórtico y mitral, resto sin alteraciones.
Electrocardiograma con ritmo sinusal, frecuencia cardíaca de 64 latidos por minuto, eje derecho y signos de sobrecarga sistólica en pared inferior compatibles con hipertrofia septal basal (figura 1). Troponina T 13,7 (normal hasta 14 ng/dL), resto de laboratorios normales.

Figura 1 Electrocardiograma con signos de sobrecarga sistólica del ventrículo izquierdo por hipertrofia septal.
El ecocardiograma transtorácico mostró MCH septal con función sistólica preservada (fracción de eyección del ventrículo izquierdo -FEVI- 68%), disfunción diastólica tipo 2 con incremento en las presiones de llenado, severa dilatación auricular izquierda, esclerosis válvula mitral e insuficiencia leve a moderada; gradiente dinámico obstructivo hacia el tracto de salida del ventrículo izquierdo de 51 mmHg; esclerosis valvular aórtica con mínima insuficiencia, ventrículo derecho sin alteraciones y dispositivo en cavidades derechas bien posicionado (figura 2).
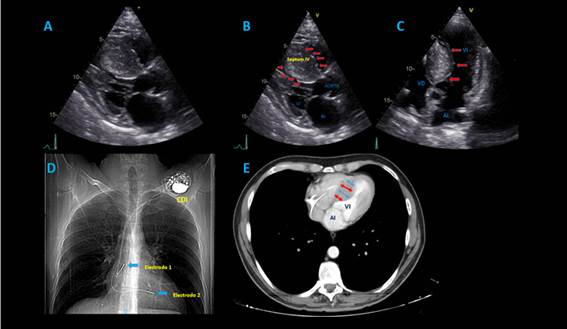
Figura 2. A-B-C: Ecocardiograma transtorácico, en imágenes multidimensionales donde se evidencia clara hipertrofia septal evaluada en vista de eje largo y de 5 cámaras. D: Rx de tórax que confirma presencia de electrodos de cardiodesfibrilador. E: Tomografía de tórax que muestra severa hipertrofia septal con reducción de volumen ventricular izquierdo.
El estudio Holter fue normal; cateterismo cardíaco sin lesiones significativas, con hallazgo incidental de puente muscular en tercio medio de la arteria descendente anterior y confirmación de hipertrofia septal asimétrica con gradiente de 83 mmHg en reposo. Telemetría de cardiodesfibrilador normal.
En junta médica se definió miectomía del septum interventricular, una vez descartado sustrato arrítmico y compromiso de fibra nerviosa pequeña de miembros inferiores como causa de pérdida del tono postural, procedimiento llevado a cabo sin complicaciones. En ecocardiograma posoperatorio hubo disminución significativa del grosor del septum interventricular y del componente obstructivo del tracto de salida del ventrículo izquierdo (figura 3). El paciente tuvo egreso hospitalario sin nueva documentación de síncopes.
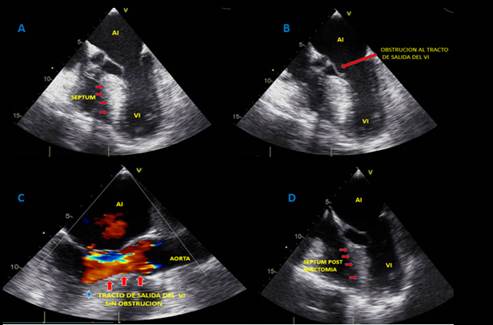
Figura 3. A-B: Ecocardiograma transesofágico, en imágenes multidimensionales donde se evidencia hipertrofia septal en vista de esófago medio con obstrucción al tracto de salida y presencia de SAM (movimiento anterior septal de válvula mitral). C: Posoperatorio de miectomía septal que demuestra liberación de la obstrucción del tracto de salida del ventrículo izquierdo. D: Vista de corrección de hipertrofia septal con reducción septal ventricular izquierdo.
Discusión
La MCH es la enfermedad genética cardíaca no sindrómica más frecuente, con transmisión autosómica dominante, sin diferencia entre sexos. Tiene una prevalencia de 1:200 a 1:500 en Estados Unidos, con un claro subdiagnóstico y subregistro1.
En Colombia, se estima una prevalencia de 2,3% y se relaciona con dolor torácico e insuficiencia mitral al momento del diagnóstico. No existe un registro que nos permita conocer la epidemiología local2.
Es causada por variantes genéticas de proteínas de la sarcómera que generan diversos grados de hipertrofia ventricular y fibrosis, lo que predispone a arritmias. Se han identificado 8 genes cuyas variantes son la cadena pesada de B-miosina (MYH7), alfatropomiosina (TPM1), troponina T cardíaca (TNNT2), proteína C ligadora de miosina cardíaca (MYBPC3), cadena ligera reguladora de miosina (MYL2), cadena ligera esencial de miosina (MYL3), troponina I cardíaca (TNNI3) y alfaactina cardíaca (ACTC1), los cuales pueden coexistir, asociándose a formas más severas1.
Variantes del gen MYBPC3 (como es el caso de nuestro paciente) se han asociado con aparición tardía y menor morbilidad3. En nuestro caso, se describió una variante probablemente patogénica que hasta el momento del estudio genético del paciente no se encontraba reportada y que podría asociarse con refractariedad. Esto puede consultarse en las bases de datos HGMD, ClinVar; de ahí la importancia de alimentar las bases de datos para una mejor interpretación.
La clínica es variable (hallazgo incidental, levemente sintomática o muerte súbita por sustrato arritmogénico mediado por fibrosis)4. Puede presentarse como falla cardíaca de predominio izquierdo con obstrucción al tracto de salida del ventrículo izquierdo (90% de pacientes con limitación funcional y refractariedad la tienen), con aumento en las presiones de llenado, regurgitación mitral, disfunción diastólica y disminución del flujo anterógrado que lleva a síncope, isquemia, disfunción microvascular, disminución de la reserva de flujo coronario y disfunción autonómica1.
El diagnóstico parte de la sospecha clínica, antecedentes y caracterización genética, se descartan primero otras causas de hipertrofia más frecuentes como la hipertensión arterial, la valvulopatía aórtica y enfermedades infiltrativas como amiloidosis5.
Es fundamental el estudio genético para definir un pronóstico, tomar decisiones más precisas y extender el estudio a familiares de primer grado6. La variante patogénica identificada en nuestro paciente se podría relacionar con formas más agresivas y refractarias al manejo, por lo que el equipo médico tuvo en cuenta este aspecto para la toma de decisiones con base en la clínica y la posible asociación entre el sustrato genético patogénico y la no respuesta al tratamiento.
En ecocardiograma, hay grosor de pared localizado o generalizado mayor de 15 mm, cuya medición es más precisa con contraste intracavitario, hipertrofia apical (diámetro mayor de 13 mm), aneurismas apicales y trombos. Es frecuente una relación de grosor septal a pared libre del ventrículo izquierdo (en diástole) mayor o igual a 1,3:1,0 y un gradiente del tracto de salida del ventrículo izquierdo mayor o igual a 30 mmHg (los síntomas son evidentes con gradiente mayor de 50 mmHg). Hay disminución del strain longitudinal y circunferencial reforzado por desacople de las fibras miocárdicas. La resonancia magnética permite una mejor caracterización tisular y descarta otras etiologías1.
En electrocardiograma se encuentran signos de hipertrofia ventricular izquierda, ondas Q patológicas no relacionadas con necrosis, ascenso o descenso del segmento ST e inversión de la onda T1.
El tratamiento se basa en betabloqueadores cardioselectivos o calcioantagonistas no dihidropiridínicos, disopiramida y diuréticos si no hay respuesta, teniendo precaución con el uso de IECA, ARA2 o calcioantagonistas dihidropiridínicos que pueden empeorar los síntomas por la obstrucción dinámica del tracto de salida del ventrículo izquierdo. La indicación de terapia de reducción septal (miectomía o ablación con alcohol) está supeditada a la clínica del paciente. Sin embargo, conocer la variante patogénica es importante, ya que puede asociarse a mala respuesta al tratamiento1.
Nuevas terapias como mavacamten (inhibidor de la ATPasa cardíaca que limita la formación de puentes cruzados de miosina-actina, lo que disminuye la contractilidad miocárdica) han mostrado impacto en la FEVI en casos de refractariedad, con FEVI mayor de 55%, clase funcional NYHA III/IV y bajo programas de estricto seguimiento basado en los resultados del ensayo EXPLORER-HCM. Sin embargo, se requieren estudios prospectivos que evalúen su real impacto en la morbilidad y mortalidad1.
Conclusión
La MCH es la enfermedad cardíaca genética más frecuente en el mundo y tiene una gran variabilidad clínica. Existen múltiples terapias farmacológicas y no farmacológicas, por lo cual es necesario el conocimiento de la variante genética patogénica, para realizar un abordaje multidisciplinario y personalizado para actuar tempranamente, en especial en casos de refractariedad como en el caso de nuestro paciente.

