











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo

 Permalink
Permalink
“No hay educación si no hay verdad que transmitir, si todo es más o menos verdad, si cada cual tiene su verdad más o menos respetable y no se puede decidir racionalmente entre tanta diversidad”. Fernando Savater, filósofo español.
Introducción
Pocos capítulos de la cardiología han generado más dudas, confusiones y controversias que el de las enfermedades del miocardio, y su nomenclatura y clasificación han constituido un desafío ininterrumpido hasta nuestros días. Un principio general para todos los sistemas de clasificación de entidades nosológicas es su condicionamiento por el nivel de información disponible en la época en que se elaboran, y las miocardiopatías (MP) son un ejemplo demostrativo. La evolución de la información en este campo ha conducido a la publicación de múltiples clasificaciones sucesivas a cargo de investigadores individuales o de consensos de expertos que representaban la posición oficial de organizaciones médicas globales, como la World Health Organization (WHO) y la International Society and Federation of Cardiology (ISFC) o de sociedades científicas del ámbito de la cardiología de particular influencia en ambos lados del Atlántico.
Luego de una prolongada etapa en la que la patología propia del músculo cardíaco fue ignorada o atribuida rutinariamente a inflamación de causa desconocida, siguió el reconocimiento de algunos patrones anatomofuncionales particulares de afectación miocárdica, aunque con escaso o nulo conocimiento relativo a su etiología, patogenia e historia natural, exceptuando quizás la frecuente comprobación de su incidencia familiar. Una muestra de tal desorientación la constituyen los casi 60 términos que se han utilizado para designar a una misma entidad, hoy conocida como miocardiopatía hipertrófica1,2 (MCH), o la denominación de la miocardiopatía dilatada (MCD) con el nombre del país desde donde procede el informe original o el de sus autores principales.
En época reciente, los notables avances en ciencias básicas (fundamentalmente la biología molecular y la genética aplicada) han develado el origen genético de muchas MP, otorgándoles finalmente identidad nosológica, y nuevas entidades han sido descritas. No obstante, a causa de su variadísima etiología, su extraordinario grado de complejidad y su frecuente superposición genotípica y fenotípica, persisten importantes incongruencias tanto conceptuales como semánticas en relación con su nomenclatura y ubicación taxonómica.
En este capítulo nos proponemos revisar la evolución de los conceptos que dieron origen a las principales clasificaciones publicadas, para posteriormente abordar el estado actual del tema, profundizando en las diferencias y aspectos debatibles de las propuestas europea y norteamericana actualmente en vigencia. Finalmente, nos introduciremos en la más reciente clasificación MOGE(S), que capitalizando los conocimientos adquiridos en las últimas tres décadas y no obstante su relativa complejidad, representa un cambio de orientación desde el mero ordenamiento taxonómico hacia la aplicación práctica en el diagnóstico clínico y la descripción detallada de la enfermedad en el paciente individual.
No es objetivo de esta revisión la descripción individual de las distintas MP, que han sido bien caracterizadas en numerosas publicaciones dedicadas a las entidades específicas.
La patología miocárdica en retrospectiva
Durante la segunda mitad del siglo XIX se mencionaba la existencia de la miocarditis crónica, concebida entonces como cualquier enfermedad del músculo cardíaco de origen no valvular y única patología propia del miocardio reconocida hasta fines del siglo, cuando comenzó a hablarse de enfermedades del músculo cardíaco con una perspectiva más amplia.
En Alemania, Krehl introdujo el concepto de enfermedad idiopática del músculo cardíaco en 18913, mientras que Josserand y Gallavardin en Francia utilizaron el término enfermedad miocárdica primaria en 1901 para la afectación miocárdica de causa desconocida, visualizando sin embargo la posibilidad de otras etiologías4.
Warren, en 1933, señaló lo inusual de la confirmación diagnóstica de miocarditis crónica en la autopsia5, con lo que la atención se centró en etiologías no inflamatorias, en especial la enfermedad coronaria (EC) y la hipertensión arterial (HTA). Advirtió dos modalidades de muerte en las MP: por deterioro gradual de una insuficiencia cardiorrespiratoria o de forma súbita.
En 1950, H. Christian destacó que un tercio de la enfermedad no inflamatoria cardíaca no puede adjudicarse a enfermedad coronaria o hipertensiva6, con lo que resurgió para este grupo de causa incierta la expresión “enfermedad miocárdica primaria”7) .
En 1956, Blankenhorn y Gall usaron el término miocarditis para designar la patología inflamatoria del músculo cardíaco, y miocardosis para otro tipo de afecciones consideradas “degenerativas”8.
El término cardiomiopatía fue utilizado por primera vez por W. Brigden en 19579 para describir la enfermedad miocárdica de origen no coronario* y la clasificó en cinco formas según su origen: congénita, infecciosa, por enfermedad del colágeno, amiloidosis, y una variedad anatómica, la fibrosis endomiocárdica, de etiología incierta. Insistió en restringir el nombre de miocarditis específicamente a la patología del miocardio de origen infeccioso y previó la progresiva reducción del grupo de las MP llamadas idiopáticas a medida que diversos agentes infecciosos causales fueran identificados.
* Si bien la angina de pecho fue descrita por Heberden en 1768, los diferentes correlatos clínicos de la oclusión coronaria comenzaron a conocerse mejor desde 1912 con las publicaciones de James Herrick10,11, por lo cual la preocupación de los clínicos al momento de diagnosticar una enfermedad propia del miocardio consistía en el siglo XIX en descartar la patología valvular, conocida desde antaño, y recién bien entrado el siglo XX involucró a la EC, de prevalencia y reconocimiento crecientes.
Con frecuencia se cita la publicación de Goodwin y Oakley de 197212 como la primera clasificación en los tres tipos morfológico-funcionales que hasta hoy utilizamos para el manejo clínico de las MP. Sin embargo, esta concepción reconoce un antecedente en el trabajo que el propio J. F. Goodwin (Figura 1), junto con otros autores, publicó en 196113, donde definió las miocardiopatías como “enfermedades del músculo cardíaco subagudas o crónicas de etiología oscura o desconocida”, y distinguió por primera vez tres formas de presentación clínica que denominó: congestiva (posteriormente dilatada), obliterativa o constrictiva (por su similitud fisiopatológica con la pericarditis constrictiva y ahora conocida como restrictiva), y obstructiva (actualmente hipertrófica), división que hasta hoy continúa siendo de utilidad metodológica para el diagnóstico clínico.

Figura 1: La primera clasificación en tres tipos básicos de comportamiento clínico de las miocardiopatías corresponde a J. F. Goodwin y colaboradores, en un artículo del cual se reproduce aquí el encabezamiento junto con la fotografía del autor principal. Goodwin utilizó un criterio fisiopatológico en base al cual dividió las miocardiopatías en congestiva, constrictiva (u obliterativa) y obstructiva, terminología que posteriormente fue modificada con la introducción de elementos morfológicos, lo que no menoscaba el valor clínico de su concepción original.
Fowler, en 1964, publicó su clasificación14, en la cual denominó como “primarias” a las MP idiopáticas, advirtiendo que en el futuro podrían identificarse dentro de ellas varias categorías etiológicas, de las cuales la miocarditis podría ser una causa muy frecuente.
Un boletín de la WHO de 196815 también designó como primarias a las MP de etiología desconocida y secundarias a las asociadas a una enfermedad sistémica o a un síndrome conocidos.
Ante este confuso panorama, diversos comités de expertos designados por instituciones mundiales de la salud o sociedades científicas de la especialidad a lo largo de las décadas siguientes intentaron ordenar la información disponible y generaron documentos con su visión del problema según el estado del conocimiento en cada época. Ello no impidió, sin embargo, que persistieran puntos oscuros, contradicciones y diferencias conceptuales que señalaremos en esta revisión.
Esfuerzos institucionales de clasificación de las miocardiopatías
Clasificación WHO/ISFC 198016
Se trata de una publicación breve y con algunas inconsistencias.
El documento define las MP como “enfermedades del músculo cardíaco de causa desconocida”, dividiéndolas, siguiendo a Goodwin, en tres patrones básicos: dilatada, hipertrófica y restrictiva (MCR), con una mínima caracterización de cada una. La (Figura 2) muestra las características anatomofuncionales básicas que permiten diferenciar dichos patrones.

Figura 2: Modelo de clasificación de las miocardiopatías según su fenotipo estructural y funcional, adoptado en el primer esfuerzo global de sistematización nosológica de las miocardiopatías a cargo de un Comité de la WHO/ISFC en 1980, criterio que la ESC mantendrá en su clasificación de 2008. Posteriormente al documento de 1980, se incorporarán otras entidades que no tienen cabida en este esquema, como la miocardiopatía arritmogénica del ventrículo derecho, la miocardiopatía no compactada y el síndrome de takotsubo. N: normal. WHO: World Health Organization; ISFC: International Society and Federation of Cardiology; ESC: European Society of Cardiology
Describe, diferenciándolo de las MP, un amplio grupo de enfermedades del miocardio causadas por o asociadas a patologías de otros sistemas, a las que denomina “enfermedades específicas del músculo cardíaco”, excluyendo expresamente la afectación del miocardio originada en la HTA sistémica o pulmonar, la EC, las valvulopatías (VP) y las cardiopatías congénitas (CC).
Estas enfermedades miocárdicas específicas -no reconocidas aquí como MP- son clasificadas según su causa en infecciosas, metabólicas, heredofamiliares, por toxicidad e hipersensibilidad, y por patologías sistémicas.
La diferenciación, sin embargo, parece más semántica que de fondo, considerando que “miocardiopatía” no significa otra cosa que “enfermedad del músculo cardíaco” sea idiopática o no, y que no resulta clara la razón por la que un desorden del músculo cardíaco originado en una patología sistémica no debiera considerarse como MP específica una vez excluidas la HTA, la EC y las VP. Este criterio sería reconsiderado en una versión posterior en 1995.
Esta clasificación de la WHO precedió al notable desarrollo de la genética aplicada a las enfermedades cardiovasculares que en las últimas tres décadas logró identificar los genes responsables de muchas MP17-19, por lo cual su asociación con enfermedad idiopática es compartible. Sin embargo, en el caso específico de la MCH el documento reconoce un patrón de herencia monogénico autosómico dominante, que conlleva implícito su origen genético -aunque sin identificación aún de los genes causales-, por lo que no sería adecuada, incluso en aquel contexto temporal, la definición general de MP como desorden de origen desconocido. De hecho, la designación que emplea “miocardiopatía hipertrófica”, utilizada en la actualidad, evita el adjetivo “idiopática” incluido en gran parte de la nomenclatura que le fue aplicada previamente. Esta incongruencia probablemente se deba a una inercia histórica del concepto por el cual una MP que no forma parte de una enfermedad identificable era considerada idiopática o primaria. Como veremos luego, las acepciones de los términos “primaria” y “secundaria” han variado en paralelo al desarrollo de la biología molecular, que ha permitido desconectar el concepto de MP con el de enfermedad de causa desconocida.
Este documento agrega además un oscuro grupo de “Miocardiopatías inclasificadas”, que no tiene cabida en ninguno de los previos, y que lo concibe como enfermedades con alteraciones menores que podrían evolucionar o no hacia la MP manifiesta, por lo que también las han denominado “Miocardiopatías latentes”.
Clasificación WHO/ISFC 1995-199620
Esta versión define las MP como “enfermedades del miocardio asociadas con disfunción cardíaca”. Abandona la distinción entre MP (entendidas en la versión previa como idiopáticas) y enfermedades específicas del músculo cardíaco, ya que la confirmación de causas tanto genéticas como adquiridas en los tres tipos morfofuncionales básicos eliminó la condición que supuestamente las diferenciaba. Ya no era aceptable que el diagnóstico de MP se condicionara al desconocimiento de su etiología.
Además de la MCD, la MCH y la MCR, incluye la miocardiopatía arritmogénica del ventrículo derecho (MAVD), inicialmente denominada displasia arritmogénica del VD y caracterizada en 198221.
Señala que la MCD puede ser tanto idiopática como de etiología familiar/genética, viral o inmune, o ambas, o tóxica/alcohólica, y que la MCH, de presentación familiar con herencia autosómica dominante, es causada por mutaciones en genes de proteínas contráctiles del sarcómero.
Acepta que algunas arritmias y trastornos de la conducción pueden constituir enfermedades miocárdicas primarias, pese a lo cual el panel de expertos elige no incluirlas entre las MP.
Razonablemente, sustituye la denominación “Enfermedades específicas del músculo cardíaco” de la versión previa por “Miocardiopatías específicas”, asociadas con desórdenes sistémicos particulares que categoriza en etiologías inflamatoria, metabólica, inmunológica, miopática, neuromuscular, tóxica y periparto, incluyendo la mayoría de ellas varias enfermedades causales.
Sorprendentemente, incluye también en este grupo de verdaderas MP la afectación miocárdica asociada con EC, HTA y VP, denominándolas “MP isquémica, hipertensiva y valvular”, respectivamente, descartadas tanto históricamente como en la versión anterior de la WHO por entender que “ampliar de esta forma el panorama etiológico podría tornar inútil la clasificación”. La condición que el panel de expertos requirió para fundamentar tal viraje conceptual fue que el deterioro de la función contráctil del ventrículo no pueda explicarse por la extensión de la EC o del daño isquémico, o resulte desproporcionado con las condiciones anormales de carga. Esta explicación resulta poco consistente, ya que si se admite que el daño miocárdico no puede adjudicarse a la enfermedad de fondo, debería considerarse como una segunda enfermedad de causa no determinada y no sería clasificable como una MP específica sino idiopática, si no hay otra causa detectable.
Como en el documento previo, continúa existiendo un subgrupo de MP no clasificadas con mejor identificación de sus integrantes, que incluyen la fibroelastosis, la miocardiopatía no compactada (MNC) descrita en 199022, la disfunción miocárdica con dilatación mínima y las MP de origen mitocondrial.
Clasificación de la American Heart Association 200623
Comienza señalando las históricas confusiones y contradicciones en la nomenclatura y clasificación de las MP, que entiende han menoscabado su comprensión y su apropiada consideración en la clínica. Aboga por mejorar la precisión del lenguaje empleado y expone la necesidad de incorporar los avances de la genética molecular en el diagnóstico etiológico y las nuevas entidades descritas en la última década, incluyendo las canalopatías iónicas con su potencial arritmogénico letal, y considera obsoleta en varios aspectos la clasificación de la WHO de 1995.
Propone una clasificación que considera en necesaria correspondencia con la era molecular de las cardiopatías y con aplicación en el diagnóstico clínico, sin pretender proporcionar una metodología o estrategia específica para este fin, admitiendo que requerirá revisión en el futuro.
Presenta una revisión crítica de la clasificación en MCD, MCH y MCR, en base a las siguientes consideraciones:
1. Mezcla criterios anatómicos (dilatación, hipertrofia) con uno funcional (restricción), con lo cual la misma enfermedad podría ubicarse en dos categorías (por ejemplo, la MCH exhibe tanto hipertrofia como restricción).
2. Algunas formas de MP pueden no tener un fenotipo estático y sufrir remodelación, pasando de una categoría a otra, como la MCH o la cardiopatía amiloidea, que eventualmente evolucionan a una MCD.
3. La identificación del sustrato genético de la enfermedad puede preceder en años a cualquier alteración morfológica o funcional objetivable, con lo cual no aplican aún las características cuestionadas.
4. En ciertas oportunidades resulta difícil distinguir formas dilatadas de no dilatadas en base a medidas cuantitativas, dado que el diámetro ventricular es una magnitud continua y hay expresiones incipientes de la enfermedad.
Concluye, en definitiva, que la clasificación morfofuncional se ha tornado de poca utilidad y que probablemente debería ser abandonada.
También expone las limitaciones propias de las clasificaciones puramente etiológicas, donde un mismo patrón anatomofuncional puede responder a múltiples etiologías, y las funcionales, dado que este enfoque tiene una aplicación fundamentalmente terapéutica, pero las estrategias de tratamiento cambian continuamente.
La AHA define las MP como “un grupo heterogéneo de enfermedades del miocardio asociadas a disfunción eléctrica y/o mecánica que usualmente (pero no invariablemente) exhiben inapropiada dilatación o hipertrofia ventricular y son debidas a una diversidad de causas que frecuentemente son genéticas. Pueden estar confinadas al corazón o ser parte de desórdenes sistémicos generalizados, y a menudo conducen a muerte cardiovascular o incapacidad progresiva relacionada con insuficiencia cardíaca”.
De esta definición y del desarrollo del documento surgen varios puntos destacables; varios de ellos novedosos con respecto a clasificaciones previas:
a) No es necesario que exista disfunción miocárdica mecánica sistólica o diastólica ni un correlato morfológico identificable en los estudios de imagen, biopsia o necropsia para considerar el diagnóstico de MP. Por lo tanto, la AHA incluye afecciones puramente eléctricas caracterizadas por un elevado riesgo de arritmias letales cuyas anomalías funcionales y estructurales se encuentran a nivel molecular, como es el caso de las canalopatías iónicas. Estas enfermedades no se detectan en un estudio de imagen, sino que se manifiestan en el electrocardiograma (ECG).
b) En oposición a la segunda clasificación de la WHO, reafirma la clásica exclusión de las afecciones del miocardio producto de HTA, EC, VP y CC, descartando, por ejemplo, la frecuente expresión “miocardiopatía isquémica”.
c) Establece una división inicial entre MP primarias y secundarias, aunque con un significado diferente del etiológico histórico, definiendo con un criterio de contexto clínico como primarias aquellas MP que se encuentran única o predominantemente confinadas al corazón, en número relativamente limitado, y como secundarias las que forman parte de una gran variedad de enfermedades sistémicas o multiorgánicas. Éstas habían sido denominadas “Enfermedades específicas del músculo cardíaco” o “Miocardiopatías específicas” por la WHO en 1980 y 1995, respectivamente. El documento reconoce que ocasionalmente esta distinción puede ser arbitraria, dado que algunas MP pueden afectar predominantemente pero no de forma exclusiva al corazón, y la decisión recae en la valoración de la importancia clínica y las consecuencias del proceso miocárdico.
d) A su vez, subclasifica las MP primarias según la naturaleza de su origen en genéticas, mixtas (genéticas y no genéticas) y adquiridas (Figura 3) y a continuación realiza una descripción de las características básicas de cada una. De esta descripción, un par de aspectos puntuales resultan relevantes:

Figura 3: Clasificación de las miocardiopatías según la American Heart Association, 2006. MCH: miocardiopatía hipertrófica; DAVD: displasia (miocardiopatía) arritmogénica del ventrículo derecho; MNC: miocardiopatía no compactada; PRKAG2: subunidad gamma-2 regulatoria de la proteinquinasa activada por el AMP; MCD: miocardiopatía dilatada; SQTL: síndrome de QT largo; SQTC: síndrome de QT corto; TVPC: taquicardia ventricular polimórfica catecolaminérgica; SUNDS: síndrome de muerte súbita inesperada nocturna.
- La MCH es descrita como una MP primaria genética (afectando única o principalmente al corazón) originada en una diversidad de mutaciones de proteínas del sarcómero codificadas por 11 genes. La distingue de una serie de MP metabólicas, infiltrativas o por acumulación de productos, neurodegenerativas o multiorgánicas (por lo tanto, secundarias) que se deben a mutaciones en genes distintos a los que codifican las proteínas sarcoméricas. Esta posición es refrendada por la Guía para el Diagnóstico y Tratamiento de la MCH publicada por la American College of Cardiology Foundation (ACCF), junto a la AHA, cinco años después de este documento24.
- Como mencionáramos previamente, esta clasificación considera como MP a las canalopatías iónicas, incluyendo los síndromes de QT largo y QT corto, el síndrome de Brugada y la taquicardia ventricular polimórfica catecolaminérgica. También se mencionan el síndrome de muerte súbita inesperada nocturna, que es el equivalente asiático del síndrome de Brugada en su base clínica y genética, y la enfermedad de Lenegre del sistema de conducción.
Clasificación de la European Society of Cardiology 200825
Señala la escasez de conocimientos sobre la etiología y la fisiopatología de las MP que caracterizó a las clasificaciones previas, aun comprendiendo que involucraban entidades diferentes.
Reconoce que la distinción entre MP primarias y secundarias, según la acepción clásica por la cual primaria se asimilaba a idiopática, ha perdido vigencia al develarse la etiología genética de muchas MP antes denominadas primarias, y renuncia a esa terminología, realizando sin embargo un comentario crítico sobre el significado diferente de esos términos que utilizó la AHA en su documento.
También desestima la posibilidad de una clasificación esencialmente genético-etiológica, argumentando que “en la práctica clínica el camino desde el diagnóstico al tratamiento no comienza en general con el hallazgo de una mutación”, y que “en caso de identificación de un gen patogénico a nivel familiar la comprobación de una enfermedad clínicamente relevante en un portador requiere demostrar un fenotipo morfológico”.
De todas formas, llega al mismo punto al dividirlas en genéticas/familiares y no genéticas/no familiares, aunque en su algoritmo de clasificación anteponga el criterio morfológico.
La ESC considera que “una miocardiopatía es un desorden en el cual el músculo cardíaco es estructural y funcionalmente anormal, en ausencia de enfermedad coronaria, hipertensión arterial, enfermedad valvular y enfermedad cardíaca congénita suficiente para causar la anormalidad miocárdica observada”.
Como se desprende de la definición, la ESC discrepa con la AHA en relación con las canalopatías, argumentando contra su interpretación como verdaderas MP, ya que requiere una alteración estructural objetivable en los estudios de imagen.
Las clasifica en fenotipos morfológico-funcionales y cada fenotipo es subclasificado en formas familiares y no familiares (Figura 4), entendiendo por familiar o genética la ocurrencia en más de un miembro de la familia de la misma enfermedad o de un fenotipo que pueda ser causado por la misma mutación y no por enfermedades cardíacas o sistémicas en las que el fenotipo clínico sea influenciado por polimorfismo genético, dado que la mayoría de las MP familiares son monogénicas.

Figura 4: Clasificación de las miocardiopatías según la European Society of Cardiology, 2008. MCH: miocardiopatía hipertrófica; MCD: miocardiopatía dilatada; MAVD: miocardiopatía arritmogénica del ventrículo derecho; MCR: miocardiopatía restrictiva.
Las MP no familiares son subdivididas en idiopáticas si no tienen causa identificable, y adquiridas si la disfunción ventricular es una complicación y no un rasgo propio de la enfermedad.
El documento afirma erróneamente que en la clasificación de la WHO/ISFC de 1995 se excluyó la disfunción ventricular producto de EC, HTA, VP o CC. Aunque estas etiologías fueron excluidas en la clasificación de 1980, en el documento de 1995 fueron incorporadas dentro del concepto de MP específicas, introduciendo un requisito diagnóstico poco compartible, previamente explicado. En la clasificación de la ESC estas condiciones clínicas quedan claramente descartadas del grupo de las MP, pero se reedita la expresión “enfermedades específicas del músculo cardíaco” (empleada en la clasificación de la WHO de 1980 con otro significado) para ubicarlas.
La (Tabla 1) sintetiza las principales diferencias conceptuales entre ambas clasificaciones.

Clasificación MOGE(S) 201326,27
Tomando nota de las limitaciones y puntos controversiales de los sistemas de clasificación precedentes, y sustentándose en una caracterización no invasiva más refinada del fenotipo y el creciente conocimiento de la etiología genética precisa de muchas MP, surgió una iniciativa de ordenamiento nosológico con un enfoque esencialmente distinto, en cuya concepción participaron cardiólogos clínicos, especialistas en insuficiencia cardíaca y trasplante, genetistas e imagenólogos tanto europeos como estadounidenses.
Esta propuesta, avalada por el Comité Científico de la World Heart Federation (WHF), está inspirada en el sistema de estadificación TNM de los tumores, en la cual se expresan con letras mayúsculas las categorías relevantes que representan la extensión de la enfermedad (Tumor primario-Nódulo linfático-Metástasis), y se define con un subíndice integrado por números y letras la situación particular del paciente en cada categoría.
En esencia, MOGE(S) no representa una verdadera clasificación, concebida como un cuadro teórico de entidades nosológicas agrupadas en categorías como los que describimos previamente, sino un sistema descriptivo de nomenclatura y notación aplicado al diagnóstico en la práctica clínica, basado en un conocimiento más profundo de las bases fisiopatológicas y genéticas de las MP. Su objetivo es precisar todos los atributos clínicos y genéticos que presenta una determinada MP en el paciente individual (fenotipo-genotipo) a fin de proveer un diagnóstico descriptivo detallado para su manejo, generando a la vez una base común con un propósito de investigación.
Los proponentes consideran que la clasificación morfológico-funcional continúa teniendo relevancia, pero la evaluación basada en el genotipo debe ser la que dirija el proceso diagnóstico y las decisiones terapéuticas en el paciente afectado y sus familiares, así como las estrategias de seguimiento.
En su publicación original, los autores definen a las MP como “desórdenes caracterizados por un miocardio morfológica y funcionalmente anormal en ausencia de cualquier otra enfermedad que sea suficiente por sí misma para causar el fenotipo observado”.
Esta definición resulta algo desconcertante, porque a diferencia de las publicaciones de la ESC y la AHA, que descartan explícitamente la EC, la VP, la HTA y la CC, con la expresión “cualquier otra enfermedad” parecería que solo incluyera las MP denominadas primarias por la AHA. Por ejemplo, la cardiopatía amiloidea no sería, según esta definición, una MP, ya que puede originar por sí misma un fenotipo de hipertrofia, restricción o eventualmente dilatación.
Dadas la complejidad clínica y etiológica de las MP y su intención de realizar una caracterización exhaustiva, resultaría muy complejo brindar una descripción completa y detallada de esta nomenclatura en la presente revisión, para cuya comprensión cabal se remite al lector a las publicaciones originales.
Básicamente se ocupa de definir cinco atributos de la MP en cuestión, donde las letras del acrónimo y los subíndices en idioma inglés tienen los siguientes significados:
M: Características Morfofuncionales que describen el fenotipo, con los subíndices MD por dilatada, MH por hipertrófica, MA por arritmogénica del VD, MR por restrictiva y MLVNC por no compactación del VI.
Se prevén además múltiples posibilidades, como superposiciones de fenotipos, atributos específicos como bloqueo auriculoventricular, síndrome de Wolf-Parkinson-White, onda épsilon, adquisición fenotípica temprana, ausencia de afectación en portador genético, etcétera, todas señaladas con sus respectivos subíndices.
O: Compromiso de Órgano(s), que puede incluir solo al corazón (O H ) o a muchos otros órganos o sistemas como músculo esquelético, (O H+M ), riñón (O H+K ), sistema nervioso, (O H+N ), hígado (O H+L ), etcétera, u O 0 en caso de portadores sanos, dado que el corazón no está aún afectado.
G: Herencia Genética o familiar , precisando si es autosómica dominante (G AD), autosómica recesiva (G AR), ligada al cromosoma X (G XL), ligada al X recesiva (G XLR), ligada al X dominante (G XLD), o transmisión matrilineal (G M). Se han previsto otros subíndices para casos esporádicos o con historia familiar negativa, desconocida o no investigada.
E: Definición Etiológica. Resulta la notación más compleja, que refiere a la causa precisa de la enfermedad, sea genética o no. Si es genética, se designa EG más el gen específico con la mutación sufrida. En el caso de una MCH, un ejemplo podría ser: E G-MYH7 (p.Arg403Glu). Puede tratarse de un no portador (EG-Neg) y hay múltiples opciones para designar presencia de más de una mutación o defectos genéticos complejos (EG-C), portadores obligados, no portadores obligados, test genético no disponible o en curso, pacientes genéticamente huérfanos, etcétera.
En caso de etiología no genética puede describirse como viral (V) agregando el virus, por ejemplo Coxsackie B3 (CB3) como E V-CB3 o Epstein-Barr virus (EBV) como E V-EBV.
Infecciones no virales se designan como E I más el agente específico si es posible, miocarditis como E M. Hay también expresiones abreviadas para enfermedades autoinmunes, amiloidosis no genética, sarcoidosis, causas tóxicas o por drogas, feocromocitoma, etcétera.
La notación de la categoría E podrá modificarse en el futuro a causa de algunas condiciones hoy excluidas de las MP o por el reconocimiento de nuevas entidades.
(S): Estadio (Stage) de insuficiencia cardíaca y clase funcional.
Se plantea como una definición opcional (de ahí el paréntesis), pero conveniente en particular para la descripción de una MP en etapa temprana. Se definen entonces el estadio A a D de insuficiencia cardíaca según la clasificación de la ACCF/AHA y la clase funcional (CF) I a IV de la New York Heart Association (NYHA). Por ejemplo, SA-I en un paciente en riesgo por ser portador genético, sin manifestaciones fenotípicas y asintomático, o SC-II en uno con alteraciones estructurales y síntomas en CF II.
Aspectos positivos que los autores señalan acerca de este sistema son la flexibilidad y la posibilidad de expansión, de modo de acompañar la evolución del conocimiento sobre la etiopatogenia de las MP. Si bien en defensa de esta propuesta se afirma que su complejidad no es tan importante como aparenta y que su manejo se hace más fluido con la experiencia, implica el uso de una extensa nómina de expresiones que resulta difícil de dominar si no se cuenta con una ayuda escrita o, tal como han elaborado y recomiendan los autores, una aplicación web que está disponible en la dirección http://moges.biomeris.com/.
Como limitaciones adicionales, se ha señalado que esta clasificación no cumple con los criterios diagnósticos de las MP en varias situaciones clínicas, y no siempre es aplicable en la práctica dada la carencia de estudios genéticos en muchos centros. Su uso podría resultar más apropiado en ámbitos de atención terciaria y particularmente en instituciones de diagnóstico e investigación genética, en los cuales el volumen de pacientes y la formación específica permitirían obtener la máxima utilidad de este sistema.
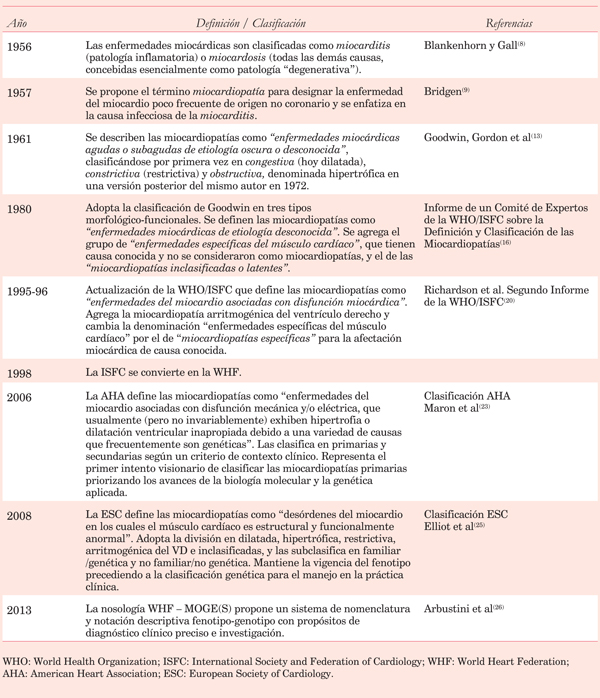
En la (Tabla 2) se resumen los puntos más destacados en la evolución del conocimiento y de los conceptos que dieron origen a las principales clasificaciones que han sido publicadas.
Tabla 2: Evolución histórica de las propuestas de clasificación de las miocardiopatías durante los últimos 60 años (modificado de Arbustini et al 27
Comentario
Analizaremos aquí solamente los aspectos confusos o controversiales de las clasificaciones de la AHA y la ESC, dado que ambas mantienen vigencia y exhiben diferencias sustanciales tanto en la definición como en los criterios para el ordenamiento taxonómico de las MP, en tanto que la clasificación MOGE(S) presenta objetivos y metodología específicos diferentes a sus predecesoras.
En primera instancia, dos aspectos básicos de coincidencia entre ambos documentos son la incorporación de los conocimientos adquiridos en el campo de la genética molecular, esenciales para su clasificación y para su manejo clínico, y la exclusión del grupo de las MP de la enfermedad miocárdica originada por HTA, EC, VP y CC, punto tratado en forma desconcertante en la clasificación de la WHO/ISFC de 1995.
Los temas controversiales incluyen:
1. El criterio de clasificación
Es el punto más crítico y objeto de agudo debate entre los defensores de una y otra posición.
La ESC utiliza un criterio primordialmente morfofuncional con el objetivo expreso de “proveer un punto de partida inicial para la investigación clínica”, concediendo importancia secundaria a la etiología familiar/genética, que aplica en un paso posterior.
Consideramos que el argumento de que la enfermedad es clínicamente relevante solo cuando se demuestra un fenotipo morfológico es difícilmente sostenible. El test genético predictivo “en cascada” entre los familiares en primer grado de un caso índice determina si se requiere o no una estrategia de seguimiento, el riesgo de arritmias letales en caso de mutaciones “malignas” o de una elevada carga familiar de muerte súbita y la posibilidad de transmisión del defecto a la descendencia28,29).
La AHA no discrimina patrones anatomofuncionales; establece una división inicial en MP primarias y secundarias y clasifica las primarias según su origen genético, adquirido o mixto, para realizar la descripción de cada entidad en una etapa posterior en forma individual, asignándolas a cada uno de esos grupos.
Una consecuencia del criterio morfológico es que reúne dentro del patrón de hipertrofia una multiplicidad de entidades muy heterogéneas, lo que ha propiciado un debate en cuanto a nomenclatura y ubicación taxonómica entre las sociedades europea y norteamericana30,31.
La ESC reconoce que históricamente el diagnóstico de MCH fue establecido ante una hipertrofia ventricular -en el sentido histopatológico del término- no explicable por condiciones de carga hemodinámica, generada en mutaciones que afectan a las proteínas del sarcómero, excluyendo enfermedades sistémicas como la amiloidosis, patologías por almacenamiento de glucógeno, enfermedades de los lisosomas, o participación miocárdica en síndromes multiorgánicos, originadas en mutaciones en otros blancos genético-moleculares.
No obstante, su posición es la de incluir a todas estas condiciones bajo el término MCH con un enfoque exclusivamente morfológico y de utilidad práctica, fundamentado en la dificultad en diferenciarlas de la verdadera hipertrofia por estudios de imagen, el dudoso rendimiento y eventual riesgo de la biopsia endomiocárdica y la necesidad de armonizar criterios con la práctica pediátrica habitual, que incluye estas enfermedades por acumulación o infiltración bajo la denominación de MCH.
Desde este marco conceptual, llega al extremo de incluir la hipertrofia por entrenamiento como MCH no familiar en su tabla 1, asimilando, al menos nominalmente, una adaptación miocárdica fisiológica, donde las condiciones de carga pueden explicar el fenotipo observado, con una entidad patológica de riesgo vital potencialmente elevado. Además, aceptando la diversidad de etiologías, mecanismos patogénicos, desórdenes asociados, tratamiento y pronóstico que esta unificación engloba, utiliza la expresión “miocardiopatías hipertróficas” en plural, con lo que se desdibujan el cuadro clínico e histopatológico de la enfermedad originalmente descrita por Teare32) y la identidad genética descubierta en los últimos años, reconocida en el documento de la WHO de 1995 y ampliamente aceptada en lo sucesivo33,34.
Establece, sin embargo, una excepción con la amiloidosis cardíaca, argumentando que aunque cumple con los criterios morfológicos de la MCH, se diferencia de ésta porque la acumulación del amiloide es intersticial y no intracelular, con lo cual no se trataría de una hipertrofia, y porque exhibe características electrocardiográficas y de imagen que sugerirían el diagnóstico diferencial.
También menciona que puede comportarse con frecuencia como restrictiva, por lo que en definitiva debería hacerse el diagnóstico diferencial entre las dos entidades.
Cabría señalar, sin embargo, que resulta muy dudosa la identificación confiable de la MP amiloidea mediante un estudio ecocardiográfico -requiriéndose una cardiorresonancia para aproximarse a este fin- y que el dato de bajo voltaje en el ECG se presenta en un porcentaje limitado de los casos35,36. Por otra parte, parece contradictorio que invocando un criterio práctico clínico-imagenológico de clasificación se utilice para diferenciarla de las demás “miocardiopatías hipertróficas” un argumento histopatológico -distribución del amiloide en el tejido miocárdico- a la vez que se reconoce la escasa aplicabilidad de la biopsia endomiocárdica en el diagnóstico clínico.
En definitiva, la ESC acepta el diagnóstico de MCH ante la simple comprobación de un aumento del espesor o de la masa ventricular -independientemente de su naturaleza y etiología- en ausencia de condiciones de carga suficientes para explicar la anormalidad observada, excluyendo la amiloidosis cardíaca, aunque reconoce que ello implica cierto grado de ambigüedad.
Sin embargo, el propio documento de 2008 incluye en su tabla 1 la cardiopatía amiloidea como MCH, y la confusión aumenta al observar que la guía de práctica clínica de la ESC de 2014 sobre el manejo de la MCH37 también incluye la amiloidosis como una de las etiologías de la MCH en el punto 4.6. bajo el subtítulo “Enfermedad infiltrativa/inflamación”.
La ACC-AHA, en cambio, solo considera como MCH una hipertrofia originada en mutaciones de proteínas sarcoméricas, diferenciándola de las demás condiciones con apariencia imagenológica similar, conocidas como fenocopias o genocopias.
Otro inconveniente del encasillamiento inmediato en un patrón morfológico ocurre con la clasificación de la miocarditis, considerada por la ESC como una MCD, cuando no siempre se traduce en este fenotipo estructural, como es el caso de la afectación exclusiva del sistema de conducción o el de la miocarditis fulminante, donde habitualmente no hay dilatación ventricular.
Como reflexión, es atendible la intención de la ESC de proveer un documento de ordenamiento nosológico con orientación clínico-práctica, pero debe aceptarse que no siempre un sistema de clasificación debe o puede oficiar de algoritmo operativo para dirigir la evaluación y el diagnóstico clínico en la consulta. Como ha expresado B. Maron: “Ninguno de los dos documentos han sido diseñados para funcionar como un libro de cocina para orientar el diagnóstico clínico; después de todo, no se trata de que los médicos atiendan al paciente junto a su cama con la clasificación de la ESC o la AHA en la mano”31.
Por otra parte, parece muy radical la afirmación de la AHA con respecto a que la clasificación anatomofuncional es una división poco útil y probablemente deba ser abandonada, ya que orienta aunque sea en forma preliminar el curso de la investigación posterior, incluso del sustrato genético.
En otras palabras, no parece adecuado descartar el criterio etiológico-genético a los efectos de una clasificación por no ser esta la primera investigación diagnóstica en la práctica clínica, ni el criterio morfológico en el estudio del paciente por la posibilidad de remodelación, superposición fenotípica o ciertos márgenes de incertidumbre. Sería recomendable disociar el objetivo de una clasificación (que es una construcción teórica) del de una estrategia de estudio en la labor clínica, evitando así renunciar a las herramientas más apropiadas para perseguir uno y otro objetivo.
Quizás lo más útil sea disponer de un documento de clasificación teórico-conceptual actualizado basado en el mejor conocimiento disponible de la etiopatogenia de las miocardiopatías, como el de la AHA, y aplicar el criterio morfofuncional de la ESC para conducir la investigación del caso individual en la práctica clínica.
2. La inclusión de las canalopatías
Otra diferencia conceptual que alimenta el debate refiere al nivel en que deben presentarse las anomalías para considerar el diagnóstico de MP. La ESC exige alteraciones estructurales objetivables en estudios de imagen, lo que excluye enfermedades puramente eléctricas como las canalopatías, que son aceptadas por la AHA, en el entendido de que representan verdaderas alteraciones estructurales y funcionales a nivel molecular en la membrana celular con modificaciones de las propiedades biofísicas de los canales iónicos, responsables de la arritmogénesis. Quizás la progresiva evolución de la medicina hacia el nivel molecular y la genómica otorguen creciente apoyo a esta interpretación en el futuro.
3. El concepto idiopática versus adquirida
Si bien la ESC claramente rechaza la identidad de los conceptos miocardiopatía e idiopática, resulta confusa su subdivisión de las MP no familiares en idiopáticas y adquiridas porque se mezclan un criterio etiológico (“idiopático”) con uno basado en el momento en que actúan el o los agentes que determinan la enfermedad (“adquirido”), dado que tanto un trastorno congénito (presente al momento del nacimiento) como uno adquirido (originado en el curso de la vida) pueden tener causa conocida o ignorada, dependiendo del nivel de información y la capacidad de investigación que se disponga. Por ejemplo, en el frecuente caso de una MCD no familiar/no genética puede no identificarse la causa y ser por lo tanto a la vez adquirida e idiopática.
El documento parece dar el nombre “adquiridas” a las MP que clasificaciones anteriores llamaban “específicas” o “secundarias”, terminología que rechaza, pero que sería una oposición más apropiada al término “idiopáticas”, en el sentido de que se generan como consecuencia o en el contexto de una patología conocida.
4. La existencia de un grupo de MP inclasificadas
Al igual que en las clasificaciones de la WHO, la ESC mantiene un limbo taxonómico de MP no clasificadas, que incluye la MNC y el síndrome de takotsubo. Lo fundamentan en su interpretación de que no resulta claro si la MNC es una MP definida o un fenotipo morfológico compartido con otras MP, y que el takotsubo implica una alteración transitoria que puede presentarse también en el curso de una hemorragia intracraneana y otros tipos de accidentes cerebrovasculares. En definitiva, más que incluir ciertas miocardiopatías en un subgrupo de difícil clasificación, la ESC plantea la duda sobre el merecimiento por derecho propio de su inclusión en una clasificación de estas enfermedades.
La AHA, en cambio, incorpora la MNC a las MP primarias de origen genético, lo que parece tener fundamento, ya que se origina frecuentemente en anomalías monogénicas o defectos cromosómicos y afecta exclusiva o predominantemente al corazón, aunque en ocasiones se presente en fenotipos que asocian a la MP desórdenes neuromusculares e inmunológicos, como el síndrome de Barth38. El síndrome de takotsubo es clasificado por la AHA como una MP primaria adquirida por estrés, aunque algunos autores han discutido si se trata efectivamente de una verdadera MP o pertenece al espectro de la patología isquémica39.
Conclusiones
Un sistema de clasificación de las enfermedades tiene el cometido de asignar nombres apropiados y ordenar jerárquicamente las distintas entidades en categorías de acuerdo con atributos (morfológicos, bioquímicos, genéticos, etcétera) compartidos o diferenciales, para facilitar su comprensión y discusión mediante la sistematización del conocimiento disponible.
La extraordinaria complejidad de las miocardiopatías y el histórico misterio en torno a su origen y fisiopatología hasta época reciente configuraron un ámbito desfavorable para su ordenamiento racional, motivo de la enorme dificultad que exhibieron casi todas las iniciativas para generar un documento conceptualmente claro, coherente y compartible por la mayoría de las sociedades científicas.
Dentro de este panorama, la clasificación de la AHA, centrada en el progresivo conocimiento de las causas genéticas de muchas miocardiopatías primarias, parece aproximarse más al modelo deseable.
Si bien no sería adecuado -como propone este documento- desconocer la utilidad de la división en patrones morfológicos para el trabajo clínico diario, tampoco es necesario que un sistema de clasificación se transforme en un manual de procedimientos para cumplir con este objetivo práctico.
La clasificación MOGE(S) apunta a un propósito diferente, y si bien constituye un valioso aporte en favor de una nomenclatura integral y una mayor precisión diagnóstica, resulta bastante compleja y su uso parece ser más apropiado en ámbitos especializados.
La discordancia de visiones y propuestas entre la AHA y la ESC constituye un obstáculo mayor para la comprensión integral de estas enfermedades, tanto en el mundo académico como para el cardiólogo práctico.
En el futuro cercano, la biología molecular y la genética aplicada continuarán aportando insumos invalorables para cumplir este objetivo. Sería deseable que esta información rindiera el máximo provecho al ordenarse en forma consensuada entre las dos principales sociedades científicas cardiológicas del mundo mediante la elaboración de un documento común.

