










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Links relacionados
Compartir

 Permalink
PermalinkRevista Uruguaya de Cardiología
versión On-line ISSN 1688-0420
Rev.Urug.Cardiol. vol.29 no.3 Montevideo dic. 2014
Artículo de revisión
CARDIOPATÍAS CONGÉNITAS
DEL ADULTO
Hipertensión arterial pulmonar
Dr. Pedro Trujillo1
1. Profesor Adjunto de la Cátedra de Cardiología, Centro Cardiovascular Universitario.
Cardiólogo Intervencionista del Servicio de Hemodinamia de Adultos del Instituto de Cardiología Integral- Médica Uruguaya.
Palabras clave:
ADULTO
CARDIOPATÍAS CONGÉNITAS-clasificación
CARDIOPATÍAS CONGÉNITAS-fisiopatología
COMPLEJO DE EISENMENGER
HIPERTENSIÓN PULMONAR-fisiopatología
HIPERTENSIÓN PULMONAR-quimioterapia
Key words:
ADULT
HEART DEFECTS, CONGENITAL-classification
HEART DEFECTS, CONGENITAL-physiopathology
EISENMENGER COMPLEX
HYPERTENSION, PULMONARY-physiopathology
HYPERTENSION, PULMONARY-drug therapy
Definición y clasificación
La circulación pulmonar normal es un circuito aislado de alto flujo y baja presión, resultado final de un proceso de evolución de los mamíferos dirigido a optimizar el intercambio gaseoso. Esta evolución se ha acompañado de una remodelación progresiva del ventrículo derecho, transformándolo en un generador de flujo, de paredes finas, incapaz de soportar bruscos incrementos de la poscarga(1).
La presión arterial pulmonar media (PAPm) normal, en reposo, es de 13 mmHg (entre 8 y 20), independientemente del sexo y de la edad. La resistencia vascular pulmonar (RVP) normal es 55 dynas·s·cm-5 (entre 12 y 100) y su correlación en unidades Wood se infiere a partir de la equivalencia una unidad Wood = 80 dynas·s·cm-5 (1).
Se define hipertensión arterial pulmonar (HP) como aquella condición hemodinámica y fisiopatológica caracterizada por un aumento de PAPm ³ 25 mmHg en reposo, estimada mediante el cateterismo cardíaco derecho (CCD) (2,3).
Existe una clasificación hemodinámica y otra clínica de la HP, ambas recientemente corregidas en el 5° Simposio Mundial de Hipertensión Pulmonar, celebrado en Niza, Francia, en febrero de 2013. La clasificación hemodinámica (tabla 1) está basada exclusivamente en la información obtenida directamente del CCD (flujo pulmonar, presión capilar pulmonar y RVP) e identifica tres grupos, HP precapilar, HP poscapilar e HP combinada (pre y pos). Aporta información conceptual y es de gran utilidad para dilucidar el o los mecanismos fisiopatológicos involucrados en la HP estudiada.

La clasificación clínica de la HP ha evolucionado en forma paralela al conocimiento patológico de las enfermedades que subyacen a este síndrome. Fue propuesta por primera vez en el año 1973 por la Oganización Mundial de la Salud en Ginebra (Suiza) y revisada sucesivamente en Evian (Francia) en 1998 y Venecia (Italia) en el año 2003. En Dana Point 2008 (California, Estados Unidos) la clasificación clínica se consolidó con algunos cambios, identificándose cinco grupos de HP con diferente mecanismo patogénico, evolución y tratamiento(4-8):
· El grupo 1 es una entidad poco frecuente con una prevalencia de 15 personas por millón de habitantes; esta HP puede ser idiopática, heredable, asociada a medicamentos o toxinas y a situaciones clínicas diversas, por ejemplo: conectivopatías, hipertensión portal, cardiopatías congénitas (CC) e infección por VIH, entre otras.
·
· El grupo 3 es la HP vinculada a la enfermedad pulmonar obstructiva crónica (EPOC), de la cual es una complicación frecuente que afecta su pronóstico.
· El grupo 4 de HP es la enfermedad tromboembólica crónica que representa entre 5% a 10% de los casos.
· El grupo 5 es un grupo heterogéneo de causa no aclarada y mecanismos desconocidos.
En Niza 2013 se hicieron algunas modificaciones en los cinco grupos mencionados que se expresan en la tabla 2(9).
Prevalencia y clasificación de las cardiopatías congénitas con HP
Se estima que de 4% a 15% de las CC desarrollarán HP y de 1% a 6% evolucionarán a síndrome de Eisenmenger. La presencia de HP tiene un impacto adverso en la calidad de vida y en la evolución de las CC(6-9).
En el Registro Nacional francés de HP, las CC asociadas a HP ocupan el segundo lugar en frecuencia, luego de la HP asociada a conectivopatías(10). Clásicamente todas las CC son consideradas causa de HP, algunas producen hipertensión pulmonar venosa, otras producen hipertensión arterial pulmonar y existe un grupo que producen HP no clasificable. Todas están representadas en la clasificación clínica general de la HP de Niza 2013, y pertenecen respectivamente a los grupos 1, 2 y 5. Las CC productoras de HP son los cortocircuitos izquierda-derecha pre y postricuspídeos así como las anastomosis confeccionadas quirúrgicamente (cirugías paliativas) que exponen a la vasculatura pulmonar a hiperflujo e hipertensión. Las CC productoras de hipertensión venosa pulmonar son las obstrucciones al tracto de salida o entrada al ventrículo izquierdo y las miocardiopatias congénitas. En el grupo 5 se incluyen: la HP segmentaria, la transposición de los grandes vasos y aquellos pacientes con HP luego de la confección de una circulación de Fontan por ventrículo único.
Existe una subclasificación fisiopatológica y anatómica descriptiva de las CC que considera el tipo y las dimensiones del defecto, la dirección del cortocircuito, la presencia de anormalidades cardíacas u extracardíacas asociadas y el estado de reparación (tabla 3) (11,12) .

Se ha propuesto también otra clasificación clínica y fisiopatológica que incluye todo el espectro de CC con HP (tabla 4) (13).
La utilidad de estas clasificaciones es permitir una detallada descripción de cada condición particular, definiendo el mecanismo fisiopatológico involucrado. Sin embargo, para el uso práctico clínico, cuatro fenotipos diferentes han sido establecidos en las CC con HP. Estos difieren en el manejo, pronóstico y respuesta al tratamiento, por lo que esta clasificación es de gran utilidad (tabla 5)(9).

Anatomía de las cardiopatías congénitas asociadas a hipertensión arterial pulmonar
La comunicación interventricular (CIV) es la CC que más frecuentemente causa HP(14). Existen múltiples tipos de CIV dependiendo de su ubicación, aunque es habitual la coexistencia de más de un defecto en el septum interventricular.
La CIV vinculada al tracto de entrada forma parte de los defectos del canal auriculoventricular (AV) y frecuentemente se asocia al síndrome de Down. Esta variante es la causa más frecuente de HP (40% de los pacientes con esta variante de CIV desarrollan HP). El tamaño de la CIV es estimado como la relación de diámetros entre la CIV y el anillo aórtico, los defectos con diámetros menores o iguales a 25% del anillo aórtico son definidos como pequeños o restrictivos. Esto limita el cortocircuito y en este escenario es menos probable el desarrollo de HP. Los defectos con diámetros mayores a 75% del anillo aórtico son definidos como grandes y la probabilidad del desarrollo de HP aumenta significativamente.
El retorno venoso pulmonar anómalo (RVPA) parcial funciona como un cortocircuito de baja presión, pretricuspídeo, que genera una sobrecarga de volumen al ventrículo derecho y a la circulación pulmonar de manera similar al comportamiento de una CIA. Estudios en autopsias demuestran que el RVPA parcial es una entidad poco frecuente, con múltiples variaciones topográficas y con una incidencia de 0,6% a 0,8% que aumenta en los pacientes con síndrome de Turner(16,17). Además de una fuerte asociación con la CIA tipo seno venoso superior, puede presentarse en un 5% a 10% de las CIA de tipo ostium secundum(18). Si bien existen múltiples reportes de casos de HP vinculados a RVPA en ausencia de otra CC, no existe una estimación de la relación entre HP y RVPA aislado.
El ductus arterioso permeable (DAP) representa un 5% a 10% de las CC y es un cortocircuito postricuspídeo que puede determinar el desarrollo de HP dependiendo del tamaño y entidad del defecto. En algunas series este cortocircuito representa el 20% de las CC que desarrollan HP(12).
La ventana aortopulmonar es una rara CC, similar al DAP, habitualmente la comunicación es amplia, sin restricción, por lo que evoluciona a síndrome de Eisenmenger si no se repara quirúrgicamente en etapas tempranas(19).
El truncus arteriosus es una CC poco frecuente caracterizada por un único vaso emergente del corazón, desde donde nacen en su porción ascendente las arterias pulmonares y las arterias coronarias. La CIV está presente en forma universal. Esta CC, si no es reparada tempranamente, evoluciona a síndrome de Eisenmenger. En pacientes adultos previamente operados de esta cardiopatía, puede existir un cortocircuito residual que sea determinante en el desarrollo de HP(19).
El ventrículo derecho con doble salida y la CIV subaórtica o subpulmonar sin obstrucción al flujo pulmonar son las CC que más frecuentemente se asocian a HP.
Las anastomosis o cortocircuitos quirúrgicos son tratamientos paliativos que se desarrollaron con el objetivo de incrementar el flujo en el circuito pulmonar en aquellas CC en las que la perfusión pulmonar estaba impedida. En las décadas de 1960 y 1970, la experiencia quirúrgica en defectos cardíacos congénitos creció y se descubrió que estas anastomosis quirúrgicas, de alto flujo y presión, comúnmente desencadenaban HP. Tres fueron las anastomosis quirúrgicas desarrolladas: Blalock Taussig y sus modificaciones, Pott y Cooley Waterston. Estas dos últimas se asocian a mayor propensión para el desarrollo de HP y por esta razón fueron abandonadas.
El desarrollo de cambios en las arterias pulmonares originados por el aumento mantenido del flujo y la presión en el circuito pulmonar es un proceso dinámico y multifactorial, con progresiva disfunción endotelial que determina vasoconstricción y remodelación del lecho vascular pulmonar(20). Los cambios tempranos en el árbol vascular pulmonar son totalmente reversibles si la cardiopatía subyacente es corregida en tiempo y forma adecuada. Si, en cambio, la cirugía correctiva se efectúa más tardíamente, luego del segundo año de vida la presión pulmonar podría disminuir sin alcanzar los valores normales(21,22). La corrección de la CC, una vez establecida la HP, puede acelerar la progresión de la enfermedad y desencadenar una falla ventricular derecha, sugiriendo que existe un punto de “no retorno”, lo que tiene implicancias clínicas y terapéuticas de relevancia.
En un pequeño subgrupo de pacientes con un mínimo cortocircuito de izquierda a derecha se puede desarrollar una enfermedad vascular pulmonar severa fuera de proporción respecto al tamaño del defecto, lo cual se explica por la existencia de una predisposición adicional (genética), que sumada al cortocircuito desencadenan HP.
Con respecto a las diferencias pronósticas de la enfermedad vascular pulmonar secundaria a las CC, se especula que tendría que ver con que en etapas tempranas de la vida el ventrículo derecho se adapta y remodela mejor para hacer frente a las altas presiones pulmonares, como lo hacía en la etapa fetal; de hecho, existen muchas similitudes entre el corazón en un síndrome de Eisenmenger y el corazón con circulación fetal(22,23). La inversión del flujo (derecha a izquierda) en el cortocircuito descomprime la hipertensión de las cavidades derechas y preserva la función del ventrículo derecho, teniendo como consecuencia el desarrollo de cianosis y otras complicaciones propias del síndrome de Eisenmenger.
El síndrome de Eisenmenger (SE) es un desorden multisistémico, resultante del desarrollo de HP severa y mantenida en una CC. Fue detallado por primera vez por Victor Eisenmenger en 1897 para describir a un paciente con disnea desde la infancia, que muere por una hemoptisis masiva, y que en la autopsia se identifica un gran defecto septal y una vasculatura pulmonar anormal. En 1958, el Dr. Paul Wood acuña el término de síndrome de Eisenmenger para caracterizar a aquellos pacientes con HP, alta RVP y grandes defectos septales(24). Posteriormente, múltiples estudios permitieron expandirlo a otras CC. La fisiopatología de este síndrome implica el aumento progresivo de la RVP e irreversibilidad de la misma, alcanzando un punto en que la presión pulmonar excede a la presión sistémica y el flujo se invierte a través del defecto congénito, pasando a ser de derecha a izquierda (figura 1).

La hipoxemia crónica y la cianosis características de este síndrome generan numerosas consecuencias: policitemia, hiperviscosidad, trombosis, gota y dolores óseos. Asimismo, este grupo de pacientes presenta complicaciones graves, cardíacas y extracardíacas. Las complicaciones cardíacas incluyen: insuficiencia cardíaca, muerte súbita, arritmias y endocarditis infecciosa. Las complicaciones extracardíacas son: accidente cerebrovascular, absceso cerebral, tromboembolismo pulmonar y hemoptisis(25). Las causas de muerte en estos pacientes, de acuerdo a una serie analizada de 188 casos durante 31 años, fueron: muerte súbita 30%, falla cardíaca 23% y hemoptisis masiva 11%(26).
Criterios de operabilidad
Un porcentaje de pacientes con CC, y en especial con cortocircuitos de izquierda a derecha, no son detectados hasta la vida adulta y son diagnosticados tardíamente, una vez que la lesión vascular pulmonar se ha desarrollado. Además, en los países en vías de desarrollo existe falta de oportunidad para la reparación quirúrgica temprana del defecto en una proporción importante, todo lo cual determina que la presencia de una CC con HP no sea un hecho infrecuente. La reparación quirúrgica en el grupo de pacientes con resistencias pulmonares altas e HP establecida es riesgosa en un doble aspecto(27). Por un lado, la mortalidad perioperatoria es alta, y en especial la “crisis de HP”, que aunque en las últimas décadas se han desarrollado estrategias de tratamiento basadas en el uso de óxido nítrico con buenos resultados, permanece como un problema primordial(28). Por otro lado, superada la etapa aguda, si las RVP permanecen altas luego de la cirugía y la HP persiste, aunque el cortocircuito esté cerrado, el pronóstico es pobre y similar al de la HP idiopática. Más aún, la sobrevida de este grupo de pacientes (cortocircuito cerrado y evolución rápida a la falla del ventrículo derecho) es menor que la de los pacientes con cortocircuito abierto y SE(29) . Todo lo cual magnifica la importancia y trascendencia de la decisión de operar en este grupo de pacientes.
· Una RVP basal indexada menor de 6 unidades Woods/m2, asociada a un índice de RP/RS menor de 0,3, sin necesidad de realizar un test de vasorreactividad, es indicativo de un buen pronóstico luego de la cirugía.
· El uso de un test de vasorreactividad, utilizando oxígeno/óxido nítrico está indicado cuando la RVP indexada está entre 6 y 9 unidades Woods/m2 y el índice de RP/RS es entre 0,3 y 0,5.
· Si bien no existe un consenso absoluto, se considerará como criterios a favor de la buena evolución posquirúrgica una disminución de 20% de las RVP indexadas y del índice de RP/RS, así como valores finales de RVP indexada menores de 6 unidades Wood/m2 y relación de la RP/RS final menor de 0,3.
Recientemente ha sido publicado en el 5º Simposio Mundial de HP una modificación y simplificación de los valores mencionados arriba que se muestran en la tabla 6(9).

Queda también de manifiesto que son necesarias herramientas más precisas y menos invasivas para la toma de decisiones de operabilidad en el grupo de pacientes límite de acuerdo al perfil hemodinámico. La biopsia de pulmón es útil, invasiva y, en definitiva muy poco práctica para el uso clínico en la toma de decisiones de operabilidad. Se ha postulado recientemente que las células endoteliales circulantes (CEC), ya reconocidas como un marcador no invasivo de disfunción, remodelación y daño vascular, podrían ser un biomarcador útil en la identificación de pacientes con alto riesgo de desarrollar HP irreversible luego de la reparación del defecto congénito(31).
Tratamiento: medidas generales
Debido a la complejidad de la condición de estos pacientes, alta mortalidad, baja prevalencia y múltiples opciones de tratamiento, se recomienda que sean tratados en centros de referencia. Las medidas generales incluyen mantener una actividad física moderada, evitando los ejercicios extenuantes y la deshidratación. El embarazo está contraindicado por el alto riesgo de muerte materno-fetal y debe realizarse profilaxis de endocarditis infecciosa. Los pacientes con SE son especialmente susceptibles a la anestesia general y a la cirugía. El oxígeno suplementario domiciliario está indicado si mejora la saturación significativamente y puede mejorar los síntomas, pero no la sobrevida. Existe un riesgo aumentado de trombosis de las arterias pulmonares así como de hemoptisis(32), por lo que los anticoagulantes están indicados exclusivamente cuando se demuestra trombosis pulmonar en ausencia de hemoptisis o si esta es leve(2). La hemoptisis relevante debe ser evaluada en el sentido de considerar la embolización selectiva de arterias bronquiales. La flebotomía debe ser evitada, excepto como tratamiento de la hiperviscosidad cuando el hematocrito supera el 65%.
Tratamiento específico de la HP
BREATHE (Bosentan Randomised Trial of Endothelin Antagonist-5) y su extensión a largo plazo son estudios multicéntricos, doble ciegos, randomizados y controlados con placebo, que demuestran el beneficio del uso de bosentán en los pacientes con SE, en términos de mejoría de la calidad de vida, capacidad de ejercicio, valores hemodinámicos y clase funcional, comparado con placebo, independientemente de la topografía del defecto septal (CIA y CIV). Por otra parte, en ambos estudios no se evidenció una disminución de la saturación arterial de oxígeno, demostrando ausencia de efectos negativos sobre el cortocircuito(33-35).
Un metaánalisis, que incluyó a 410 pacientes, reafirmó la mejoría clínica y hemodinámica con el uso de bosentán, observándose un aumento significativo de las enzimas hepáticas, aunque no se demostraron efectos colaterales severos(36).
Existe un déficit de estudios clínicos, randomizados y controlados para el uso de otras terapias específicas de la HP; los datos disponibles de estudios pequeños señalan el beneficio del uso prolongado de epoprostenol, demostrando mejoría en la calidad de vida, clase funcional y parámetros hemodinámicos(37).
El tratamiento con sildenafil mejora la capacidad de ejercicio, el escore de disnea, la clase funcional, la calidad de vida y los parámetros hemodinámicos de los pacientes con HP asociada a CC y en el SE(38,39).
La utilidad de las terapias específicas en la estrategia treat to close, para reducir la RVP y llegar a la operabilidad en pacientes con HP asociada a CC, ha sido aceptada e incluida en las guías de Grown Up con una débil recomendación IIB nivel de evidencia C. Esto incluye la CIA o CIV con QP/QS >1,5, PVR menor o igual a 5 unidades Wood y PAP-PVR menor a los dos tercios de los valores sistémicos, en condiciones basales o luego de un test vasodilatador(32).
El trasplante corazón-pulmón es una opción potencial de tratamiento en este grupo de pacientes, pero está limitado por la escasez y falta de especificidad de órganos donantes.
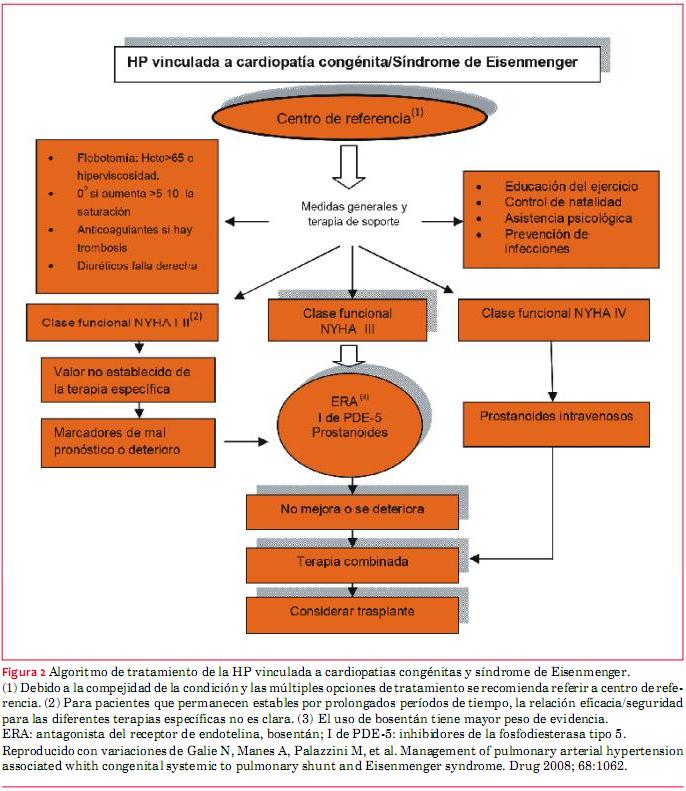
En la figura 2 se esquematiza en forma global el tratamiento de los pacientes con HP asociada a CC y al SE.
Bibliografía
1. Naeije R. Physiology of the pulmonary circulation and the right heart. Curr Hypertens Rep 2013; 15(6):623-3.1
2. Galiè N, Hoeper M, Humbert M, Torbicki A, Vachiery JL, Barberá JA, et al. Guía de práctica clínica para el diagnostico y tratamiento de la hipertensión pulmonar. Versión corregida el 27 de abril de 2011. Rev Esp Cardiol 2009; 62(12): 1464.e1-e58.
3. Chatterjee K, De Marco T, Alpert JS. Pulmonary hypertension. hemodynamic diagnosis and management. Arch Intern Med 2002;162(17):1925-33.
4. Barst R, Rubin LJ. World Symposium on Pulmonary Hypertension, 4th. Dana Point, California, feb. 11-14, 2008.
5. Marelli AJ, Therrien J, Mackie AS, Ionescu-Ittu R, Pilote L. Planning de specialized of adult congenital heart disease patients: from numbers to guidelines; an epidemiologic approach. Am Heart J 2009; 157(1):1-8.
6. Duffels MG, Engelfriet PM, Berger RM, van Loon RL, Hoendermis E, Vriend JW, et al. Pulmonary arterial hypertension in congenital heart disease: an epidemiologic perspective from a Dutch registry. Int J Cardiol 2007; 120(2):198–204.
7. Lowe BS, Therrien J, Ionescu-Ittu R, Pilote L, Martucci G,Marelli AJ. Diagnosis of pulmonary hypertension in the congenital heart disease adult population impact on outcomes. J Am Coll Cardiol 2011; 26: 538–46.
8. Engelfriet PM, Duffels MG, Möller T, Boersma E, Tijssen JG, Thaulow E, et al. Pulmonary arterial hypertension in adults born with a heart septal defect: the Euro Heart Survey on adult congenital heart disease. Heart 2007; 93(6): 682-7.
9. Simonneau G, Gatzoulis M, Adatia I, Celermajer D, Denton C, Ghofrani A, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 2013; 62(25 Suppl):D34-41.
10. Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, Gressin V, et al. Pulmonary arterial hypertension in France: results from a national registry. Am J Respir Crit Care Med 2006; 173(9):1023–30.
11. Simonneau G, Robbins IM, Beghetti M, Channick RN, Delcroix M, Denton CP, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 2009; 54(1Suppl): S43-54.
12. Adatia I, Kothari SS, Feinstein JA. Pulmonary hypertension associated with congenital heart disease: pulmonary vascular disease: the global perspective. Chest 2010; 137(6 Suppl):52S-61S.
13. Schulze-Neick I, Beghetti M. Classifying pulmonary hypertension in the setting of the congenitally malformed heart– cleaning up a dog’s dinner. Cardiol Young 2008; 18(1):22-5.
14. Steele PM, Fuster V, Cohen M, Ritter DG, Mc-Goon DC. Isolated atrial septal defect with pulmonary vascular obstructive disease–long-term follow-up and prediction of outcome after surgical correction. Circulation 1987; 76(5):1037-42.
15. Webb G, Gatzoulis MA. Atrial septal defects in the adult: recent progress and overview. Circulation. 2006; 114(15):1645-53.
16. Hoffman JI, Kaplan S. The incidence of congenital heart disease. J Am Coll Cardiol 2002; 39(12): 1890-900.
17. Healey JE Jr. An anatomic survey of anomalous pulmonary veins: their clinical significance. J Thorac Surg 1952; 23(5):433-44.
18. Ellis AR. Partial anomalous pulmonary venous connections and the Scimitar Syndrome. In: Gatzoulis MA, Webb GD, Daubeney PEF, eds. Diagnosis and Management of Adult Congenital Heart Disease. 2nd ed. Philadelphia, PA: Saunders, 2011.
19. Kiefer TL, Bashore T. Anatomy of Congenital Heart Disease Lesions Associated With Pulmonary Arterial Hypertension. Adv Pulm Hipertens 166-170. Disponible en: http://www.phaonlineuniv.org/journal.
20. Humbert M, Sitbon O, Simonneau G. Treatment of pulmonary arterial hypertension. N Engl J Med 2004; 351: 1425–1436.
21. Balint OH, Samman A, Haberer K, Tobe L, McLaughlin P, Siu SC, et al. Outcomes in patients withpulmonary hypertension undergoing percutaneous atrial septal defect closure. Heart 2008; 94(9):1189-93.
22. Hopkins WE. The remarkable right ventricle of patients withEisenmenger syndrome. Coron Artery Dis 2005; 16: 19-25.
23. Rosenzweig EB, Barst RJ. Congenital heart disease and pulmonary hypertension: pharmacology and feasibility of late surgery. Prog Cardiovasc Dis 2012;55(2):128-33.
24. Hopkins WE, Waggoner AD. Severe pulmonary hypertensionwithout right ventricular failure: the unique hearts of patients with Eisenmenger syndrome. Am J Cardiol 2002; 89:34-8.
25. Wood P. The Eisenmenger syndrome or pulmonary hypertension with reversal shunt. Br Med J 1958; 2(5099): 755-62.
26. DeFilippis AP, Law K, Curtin S, Eckman JR. Blood is thicker than water: the management of hyperviscosity in adults with cyanotic heart disease. Cardiol Rev 2007; 15(1):31-4.
27. Daliento L, Somerville J, Presbitero P, Menti L, Brach-Prever S, Rizzoli G, et al. Eisenmenger syndrome factors relating to deterioration and death. Eur HeartJ 1998; 19(12):1845-55.
28. Haworth SG, Hislop AA. Treatment and survival in children with pulmonary arterial hypertension: the UK Pulmonary Hypertension Service for Children 2001-2006. Heart. 2009; 95:312-7.
29. Bando K, Turrentine MW, Sharp TG, Sekine Y, AufieroTX, Sun K, et al. Pulmonary hypertension after operations for congenital heart disease: analysis of risk factors and management. J Thorac Cardiovasc Surg 1996; 112: 1600-7.
30. Hopkins WE, Ochoa LL, Richardson GW, Trulock EP. Comparison of the hemodynamics and survival of adults with severe primary pulmonary hypertension or Eisenmenger syndrome. J Heart Lung Transplant 1996;15: 100-5.
31. Lopes AA, O’Leary PW. Measurement, interpretation and use of haemodynamic parameters in pulmonary hypertension associated with congenital cardiac disease. Cardiol Young 2009; 19:431-5.
32. Smadja DM, Gaussem P, Mauge L, Israel-Biet D, Dignat-George F, Peyrard S, et al. Circulating endothelial cells: a new candidate biomarker of irreversible pulmonary hypertension secondary to congenital heart disease. Circulation 2009; 119: 374-8.
33. Baumgartner H, Bonhoeffer P, De Groot N, de Haan F, Deanfield JE, Galie N, et al. ESC guidelines for the management of grown-up congenital heart disease (new version 2010). Eur Heart J 2010; 31(23): 2915-57.
34. Galiè N, Beghetti M, Gatzoulis MA, Granton J, Berger RM, Lauer A, et al. Bosentan therapy in patients with Eisenmenger syndrome: a multicenter, double-blind, randomized, placebo-controlled study. Circulation 2006; 114(1): 48-54.
35. Gatzoulis MA, Beghetti M, Galiè N, Granton J, Berger RM, Lauer A, et al. Longer-term bosentan therapy improves functional capacity in Eisenmenger syndrome: results of the BREATHE-5 open-label extension study. Int J Cardiol 2008; 127(1):27-32.
36. Berger RM, Beghetti M, Galiè N, Gatzoulis MA, Granton J, Lauer A, et al. Atrial septal defects versus ventricular septal defects in BREATHE-5, a placebo-controlled study of pulmonary arterial hypertension related to Eisenmenger’s syndrome: a subgroup analysis. Int J Cardiol 2010; 144(3): 373-8.
37. Lee YH, Song GG. Meta-analysis of randomized controlled trials of bosentan for treatment of pulmonary arterial hypertension. Korean J Intern Med 2013; 28(6):701-7.
38. Rosenzweig EB, Kerstein D, Barst RJ. Long-term prostacyclin for pulmonary hypertension with associated congenital heart defects. Circulation 1999; 99: 1858-65.
39. Zhang ZN, Jiang X, Zhang R, Wu BX, Zhao QH, Wang Y, et al. Oral sildenafil treatment for Eisenmenger syndrome: a prospective, open-label, multicentre study. Heart 2011; 97: 1876-81.
40. Chau EM, Fan KY, Chow WH. Effects of chronic sildenafil in patients with Eisenmenger syndrome versus idiopathic pulmonary arterial hypertension. Int J Cardiol 2007; 120: 301-5.
41. D’Alto M, Romeo E, Argiento P, Sarubbi B, Santoro G, Grimaldi N, et al. Bosentan-sildenafil association in patients with congenital heart disease-related pulmonary arterial hypertension and Eisenmenger physiology. Int J Cardiol 2012; 155(3): 378-82.
42. Iversen K, Jensen AS, Jensen TV Vejlstrup NG, Søndergaard L. Combination therapy with bosentan and sildenafil in Eisenmenger syndrome: a randomized, placebo-controlled, double-blinded trial. Eur Heart J 2010; 31(9):1124-31.


{kind=link}
{kind=link}