










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Links relacionados
Compartir

 Permalink
PermalinkArchivos de Pediatría del Uruguay
versión On-line ISSN 1688-1249
Arch. Pediatr. Urug. vol.77 no.4 Montevideo dic. 2006
CASO CLÍNICO
Arch Pediatr Urug 2006; 77(4)
Déficit de succínico semialdehído deshidrogenasa.
Primer caso de aciduria 4 OH butírica en Uruguay
Dres. María Eugenia Russi 1, Aída Lemes 2, Gabriel González 3, Patrizia Malaspina 4
1. Clínica pediátrica “C” Profesora Ivonne Rubio. Centro Hospitalario Pereira Rossell.2. Hospital Italiano. Instituto de Genética Médica.
3. Neuropediatra. Centro Hospitalario Pereira Rossell.
4. Hospital Italiano. Departamento de Biología de la Universidad Tor Vergara. Roma, Italia.
Fecha de recibido: 7 de diciembre de 2006.
Fecha aprobado: 22 de diciembre de 2006.
Resumen
El déficit de succínico semialdehído deshidrogenasa es una enfermedad rara cuya alteración en la vía catabólica del ácido aminobutírico (principal neurotransmisor del sistema nervioso central) impide el paso de ácido succínico semialdehído a ácido succínico, condicionando un aumento de ácido 4 hidroxibutírico en líquido cefalorraquídeo y plasma, lo que permite la detección de la enfermedad.
La clínica es muy inespecífica, pudiendo presentar desde retraso psicomotor, hipotonía, convulsiones y ataxia no progresiva, hasta trastornos de conducta con crisis de ansiedad y rasgos autistas.
Los hallazgos a nivel electroencefalográfico incluyen enlentecimiento del ritmo de base, con descargas epileptiformes focales o generalizadas. En la neuroimagen, en tanto, pueden hallarse frecuentemente hiperintensidades bilaterales y simétricas en T2, a nivel del núcleo pálido.
Si bien el tratamiento con vigabatrina teóricamente inhibe la formación de succínico semialdehído con la consecuente reducción de los niveles de ácido 4 hidroxibutírico, no existe al momento actual un tratamiento efectivo para todos los casos.
El principal objetivo de este artículo es presentar un paciente con deficiencia de succínico semialdehído deshidrogenasa, debido a su excepcionalidad, con solamente 350 casos identificados en el mundo, siendo el primer caso diagnosticado en Uruguay.
Palabras clave:
ÁCIDO GAMMAAMINOBUTÍRICO -metabolismo
VIGABATRIN-uso terapéutico
ENFERMEDADES METABÓLICAS
ENFERMEDADES DEL SISTEMA NERVIOSO
Summary
Succinic semialdehyde dehydrogenase deficiency is a rare disorder of the catabolic pathway of gamma-aminobutyric acid, the major central nervous system inhibitory neurotransmitter. Because of such deficiency, transamination of gamma-aminobutyric acid to succinic semialdehyde is shunted towards the production of 4-hydroxybutyric acid, a neurotoxic metabolite which becomes abundant in physiologic fluids which allows the detection of the disorder.
Clinical features are not specific and consist of psychomotor retardation, seizures, hypotonia and non progressive ataxia.
Electroencephalographic finding include background slowing and generalized or focal epileptiform discharges. Magnetic resonance imaging reveals in most of the cases, increased T2-weighted signal of the globus pallidi bilaterally and symmetrically.
Eventhough vigabatrin should theoretically inhibit the formation of succinic semialdehyde and therefore 4-hydroxybutyric acid, to date there is no effective treatment for succinic semialdehyde dehydrogenase deficiency. Due to its uniqueness, the main objective of this article is to present a patient with succinic semialdehyde dehydrogenase deficiency, with only 350 cases identified worldwide.
Key words:
GAMMA-AMINOBUTYRIC ACID-metabolism
VIGABATRIN-therapeutic use
METABOLIC DISEASES
NERVOUS SYSTEM DISEASES
Introducción
Los trastornos del metabolismo de los neurotransmisores son un grupo de afecciones neurometabólicas de reciente descripción, que suelen presentarse en la edad pediátrica. Actualmente se reconocen dos grandes grupos: los trastornos en el metabolismo de las monoaminas y los trastornos del metabolismo del ácido gamma-aminobutírico (GABA). Dentro de estos últimos son cuatro las alteraciones conocidas hasta el momento: la epilepsia dependiente de piridoxina, la deficiencia de GABA transaminasa, la homocarnosinosis y el déficit de succínico semialdehído deshidrogenasa (SSADH) o aciduria 4-OH-butírica, cuya primera descripción fue realizada por Jakobs en 1981 y posteriormente por Gibson en 1983 (1).
La deficiencia de SSADH (Mckusick 271980) es una enfermedad poco frecuente, de herencia autosómica recesiva, cuyo gen mapea en el brazo corto del cromosoma 6 (6p22), con más de 47 mutaciones descritas en la actualidad (2).
Constituye el primer defecto del metabolismo del GABA descrito en humanos (3).
En ausencia de la enzima no se produce el paso enzimático de ácido succínico semialdehído a ácido succínico, lo cual condiciona un aumento de ácido 4-OH-butírico (4-HBA) en líquido cefalorraquídeo, plasma y orina. La subsiguiente acumulación de este metabolito endógeno con demostradas propiedades neurofarmacológicas y efectos neuromoduladores (agente neurotóxico), parece ser en parte el responsable de las manifestaciones clínicas encontradas en estos pacientes (4).
La presentación clínica es extremadamente heterogénea, predominando las manifestaciones neurológicas como retraso psicomotor, convulsiones e hipotonía; hecho este que muchas veces dificulta el diagnóstico pudiéndolo confundir con encefalopatías secuelares no progresivas prenatales (4,5).
Según la bibliografía existen más de 350 casos identificados en el mundo, con no más de 150 reportados, la mayoría de los cuales proceden de Eurasia y América del Norte, con una significativa proporción de consanguinidad (37%) (4).
Hasta el momento actual, solamente dos pacientes con déficit de SSADH han sido identificados en América del Sur (Argentina) (6). Esto seguramente se deba a un subdiagnóstico de la enfermedad en las Américas, y probablemente refleje un mejor screening metabólico en la población pediátrica de la región europea.
El objetivo de este artículo es presentar los hallazgos clínico-genéticos encontrados en un paciente con déficit de SSADH, siendo el primer caso reportado en Uruguay.
Caso clínico
FS, 7 meses, sexo masculino, raza blanca, padres no consanguíneos, sanos, de herencia española. Producto de segunda gesta, embarazo y parto normales, con un peso al nacer de 3.360 gramos, talla 51 cm y perímetro craneano de 34 cm, sin patología perinatal. Buen crecimiento, correctamente inmunizado.
Retraso global del desarrollo más evidente en áreas motoras, en asociación con hipotonía, letargia y alteraciones del sueño nocturno, presentes desde el primer trimestre de vida, sin progresividad. Ocasionalmente orinas de coloración anormal (verde oscuro) y movimientos anormales de los miembros superiores.
Al examen físico se destaca: peso y talla ambos en percentil 50, perímetro craneano en percentil 90, vigilia e interacción social fluctuantes con escaso contacto ocular, somnoliento por momentos. Hipotonía global no parética, evidenciada con la maniobra “pull to sit” (figura 1) con hiporreflexia rotuliana y fuerzas conservadas.

Estereotipías manuales. Resto del examen normal.
Valoración metabólica general y del medio interno con glicemia, ionograma, ácido láctico, amonio, ácido úrico, gasometría venosa; funcional y enzimograma hepáticos, función renal, examen de orina y hormonas tiroideas normales.
Valoración oftalmológica y fondo de ojo normal.
Electroencefalograma en vigilia y sueño normales.
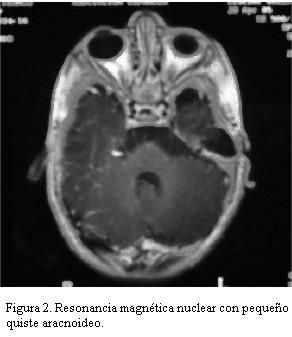
Resonancia magnética nuclear de cráneo que evidencia leve dilatación de los espacios subaracnoideos a predominio bifrontal e interhemisférico así como también del sistema ventricular, con pequeño quiste aracnoideo (figuras 2 y 3). No aumento en la señal en T2 a nivel del globo pálido, núcleo dentado cerebelar y sustancia blanca.

Estudio neurometabólico: perfil de ácidos orgánicos en orina que objetiva un aumento de los ácidos 4 hidroxibutírico y 3,4 dihidroxibutírico, así como también del 2 deoxi 1-4 tetrano lactona, el 4,5 dihidroxi hexónico lactona treo. Perfil de acilcarnitinas en sangre normal.
El análisis molecular del gen SSADH mostró una mutación 1226 G>A (missense mutation).
Con diagnóstico de déficit de SSADH, se inicia tratamiento con vigabatrina en dosis crecientes hasta alcanzar los 50 mg/kg/día y estimulación psicomotriz temprana.
En la evolución desaparecen los episodios de letargia, mejora la interacción ocular, así como los trastornos del sueño y el tono muscular, logrando la sedestación a los 8 meses de vida.
Discusión
El déficit de SSADH es una enfermedad de expresión clínica variable e inespecífica, pudiendo presentar en sus inicios moderado a severo retardo del desarrollo motor con hipotonía global y reflejos conservados (hipo o hiperrreflexia), y posterior retraso en la manipulación de objetos, con movimientos coreoatetósicos y ataxia truncal (3-5).
Si bien el retraso del desarrollo, la hipotonía y el retardo mental constituyen las características clínicas más constantes del déficit de SSADH (74 a 78%) aproximadamente el 50% de los pacientes presentan trastornos de conducta, ataxia y convulsiones; pudiendo ser éstas tónico-clónicas, mioclónicas o crisis de ausencias (4).
La expresión neurocognitiva de la enfermedad varía con la edad cronológica y la etapa del desarrollo en la que se encuentre el niño. Así, el curso clínico de la deficiencia cognitiva comienza en estadios tempranos con síntomas neurológicos como hipotonía y retraso del desarrollo, como se observa en nuestro paciente. A medida que el niño crece pueden aparecer otros síntomas, asociando retraso en la adquisición del lenguaje (por pobre habilidad fonatoria en presencia de agnosia verbal) con trastornos del comportamiento tales como agresividad, hiperquinesia, alucinaciones visuales o auditivas y conductas obsesivo-compulsivas; pudiendo encontrar en este amplio rango de trastornos conductuales un retardo psíquico global con rasgos autistas y retraso mental (7,8). Resulta interesante relacionar este espectro de trastornos de conciencia y disturbios del sueño encontrados (letargia, somnolencia diurna, insomnio nocturno, narcolepsia) con las concentraciones elevadas de 4-HBA evidenciadas en los cerebros de estos pacientes, teniendo en cuenta los efectos sedativos de este metabolito que han llevado a su uso en la práctica clínica (7,9).
No solamente responsables de las alteraciones conductuales antes descritas, la excesiva cantidad de 4-HBA en orina podría explicar la gran variabilidad de colores encontrados por otros autores (que van desde el verde oscuro hasta marrón rojizo) y espontáneamente relatado por la madre de nuestro paciente (10,11). Asimismo se ha especulado con la posibilidad de que la similitud estructural del GABA (precursor porfirínico) con el 4-HBA, sea la responsable de la cantidad excesiva de porfirina detectada en un paciente, en el cual se arribó al diagnóstico erróneo de porfiria (10). Es por ello que se puede arribar a un resultado falso positivo, al producir el 4-HBA una interferencia en el test de screening de las porfirias, elemento a tener en cuenta al momento del diagnóstico.
Considerar este amplio abanico de signos y síntomas en etapas tempranas de la vida como expresión de encefalopatías prenatales no progresivas (tales como la parálisis cerebral y los síndromes genéticos) puede llevar a confundir y retrasar el diagnóstico de esta entidad, cuyo amplio rango fenotípico contribuye en gran medida a que ello suceda. No olvidemos que estos pacientes no suelen presentarse con hipoglicemia, acidosis metabólica, hiperamoniemia y episodios de vómitos, como sí lo hacen otros pacientes con errores innatos del metabolismo contribuyendo, en gran medida, a que exista un subdiagnóstico de esta enfermedad.
En cuanto a las alteraciones neuroimagenológicas, éstas se pueden encontrar según las diferentes series en 22% a 69% de los casos, consistiendo en hiperintensidades en T2 bilaterales y simétricas a nivel del núcleo pálido, sustancia blanca y cerebelo (4,5). Otros hallazgos estructurales pueden incluir atrofia cerebro-cerebelar, a predominio vermiano (4). El hecho de que ninguna de ellas haya estado presente en nuestro paciente nos hacen especular sobre la posibilidad de que ello se deba a un diagnóstico temprano de la enfermedad.
Entretanto el trazado electroencefalográfico es anormal en aproximadamente el 50% de los casos, pudiéndose encontrar desde un enlentecimiento basal hasta descargas focales y generalizadas (4).
Para el diagnóstico bioquímico contamos con un hallazgo característico como la elevación del 4-HBA (elevaciones de 200 a 800 veces lo normal) en una muestra de orina, pudiéndose identificar otros metabolitos indicadores tanto de la beta oxidación del mismo (ácido 3,4 dihidroxibutírico y el 3-oxo-4-hidroxibutírico y glicólico) como de la alfa oxidación (ácidos 2,4 dihidroxibutírico y 3-hidroxipropiónico). Dado que el 4-HBA es muy volátil pudiendo eludir su cuantificación, es preferible el uso de la espectrometría de masas o eventualmente la detección con isótopos marcados, al examen simple de ácidos orgánicos por cromatografía de gases (5). Actualmente es posible el diagnóstico prenatal, mediante la determinación del 4-HBA en líquido amniótico y/o la biopsia de las vellosidades coriónicas (3,12).
El tratamiento con vigabatrina fue introducido como tratamiento específico del déficit de SSADH por primera vez por Jaecken y colaboradores, basado en su capacidad inhibitoria sobre la GABA transaminasa (13). Su efecto clínico beneficioso radica en la disminución de la concentración en líquido cefalorraquídeo del 4-HBA y del GABA, con la consiguiente mejoría -aunque en grado variable- del cuadro clínico (4,5,14,15).
Hecho este de interés, que ha llevado a Howells y colaboradores a postular que esta variabilidad en sus efectos podría responder a una limitada inhibición de las GABA transaminasas presentes en los órganos periféricos, con el consiguiente aumento en la producción de 4-HBA, su posterior pasaje de la barrera hematoencefálica, actuando así en detrimento de la eficacia terapéutica de la vigabatrina (5). Sin embargo se sabe que las dosis de vigabatrina utilizadas deben ser bajas (entre 25 a 50 mg/kg/día) dado el efecto deletéreo que pueden tener dosis mayores (75 a 100 mg/kg/día), habiéndose comprobado cambios electroencefalográficos y actividad epileptógena en estos casos (5,13).
Otras alternativas terapéuticas implementadas con éxito variable en estos pacientes incluyen el uso de la lamotrigina (4). El ácido valproico, por otro lado, estaría proscripto debido a una eventual inhibición de la actividad enzimática residual de la succinil semialdehído deshidrogenasa que puede estar presente en estos pacientes (4,15).
Un hallazgo importante a destacar es la dramática mejoría del tono muscular en aquellos pacientes suplementados con L–carnitina, cuando sus niveles plasmáticos se demuestran bajos (5).
Conclusiones
Si bien el déficit de SSADH es una patología clínicamente reconocida en las dos últimas décadas, nuestro reconocimiento de la misma dista mucho de ser el esperado, existiendo un claro subdiagnóstico de esta entidad a nivel mundial, producto de un amplio rango de variabilidad fenotípica. Muchos aspectos de la fisiopatología permanecen poco claros, habiéndose abierto una luz de esperanza en la aplicación de modelos animales al estudio de esta enfermedad, que en un futuro cercano puedan ampliar nuestros conocimientos. Entretanto creemos de vital importancia descartar esta enfermedad ante todo niño con retraso global del desarrollo e hipotonía global de causa no aclarada, justificando así la realización de ácidos orgánicos en orina.
Referencias bibliográficas
1. Gibson KM, Sweetman L, Nyhan WL, Jakobs C, Rating D, Siemens H, et al. Succinic semialdehyde dehydrogenase deficiency: an inborn error of gamma-aminobutyric acid metabolism. Clin Chim Acta 1983; 133(1): 33-42.
2. Gibson KM, Jakobs C. Disorders of beta -and gamma-amino acids in free and peptide-linked forms. In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The metabolic and molecular bases of inherited disease. 8 ed. New York: McGraw-Hill; 2001: 2079-105.
3. Sanjurjo P, Balldelou A, Fernandez L. Enfermedades hereditarias del metabolismo de los neurotransmisores. En: Diagnóstico y tratamiento de las enfermedades metabólicas hereditarias. Barcelona: Ergon, 2001: 547-58.
4. Pearl P, Novotny E, Acosta M, Jakobs C, Gibson KM. Succinic semialdehyde dehydrogenase deficiency in children and adults. Ann Neurol 2003; 54(6): S73-S80.
5. Gibson KM, Christensen E, Jakobs C, Fowler B, Clarke M, Hammersen G, et al. The clinical phenotype of succinic semialdehyde dehydrogenase deficiency (4-hidroxybutyric aciduria): case reports of 23 new patients. Pediatrics 1997; 99(4): 567-74.
6. Bluvstein J, Szlago M, Jorge L, Rugilo C, Chamoles N. Succinic semialdehyde dehydrogenase deficiency: case report (Abstract). Paper presented at the 4th Latin American Congress of Inborn Metabolism Errors; 2003; Misiones, Argentina.
7. Phillippe A, Deron J, Geneviéve D, Gibson KM, Rabier D, Munnich A. Neurodevelopmental pattern of succinic semialdehyde dehydrogenase. Dev Med Child Neurol 2004; 46: 564-8.
8. Gibson KM, Grupta M, Pearl LP, Tuchman M, Vezina G, Carter Snead III O, et al. Significant behavioral disturbances in succinic semialdehyde dehydrogenase (SSADH) deficiency (Gamma-hydroxibutyric aciduria). Biol Psychiatry 2003; 54: 763-8.
9. Wolf NI, Hass D, Hoffmann GF, Jakobs C, Salomons GS, Wevers RA, et al. Sedation with 4-hydroxybutyric acid: A potential pitfall in the diagnosis of SSADH deficiency. J Inherit Metab Dis 2004; 27: 291-3.
10. Gibson KM, Sweetman L, Kozich V, Pijackova A, Tscharre a, Cortez A, et al. Unusual enzyme findings in five patients with metabolic profiles suggestive of succinic semialdehyde dehydrogenase deficiency (4-hydroxibutyric aciduria). J Inherit Metab Dis 1998; 21: 255-61.
11. Gordon N. Succinic semaildehyde dehydrogenase deficiency (SSADH) (4-hydroxibutyric aciduria). Eur J Paediatr Neurol 2004; 8: 261-5.
12. Hogema BM, Akaboshi S, Taylor M, Salomons GS, Jakobs C, Schutgens RB, et al. Prenatal diagnosis of succinic semialdehyde dehydrogenase deficiency: increased accuracy employing DNA, enzyme, and metabolite analyses. Mol Genet Metab 2001; 72(3): 218-22.
13. Matern D, Lehnert W, Gibson KM, Korinthenberg R. Seizures in a boy with succinic semialdehyde dehydrogenase deficiency treated with Vigabatrin (gamma-vinyl-GABA). J Inher Metab Dis 1996; 19: 313-28.
14. Gropman A. Vigabatrin and newer interventions in succinic semialdehyde dehydrogenase deficiency. Ann Neurol 2003; 54(6): S66-S72.
15. Pearl PL, Gibson KM, Acosta MT, Vezina LG, Theodore WH, Rogawski MA, et al. Clinical spectrum of succinic semialdehyde dehydrogenase deficiency. Neurology 2003; 60: 1413-7.
Correspondencia: Dra. María Eugenia Russi.
Departamento de Pediatría, Hospital Pereira Rossell. República Argentina 1815, Montevideo, Uruguay.
E-mail: russidel@chasque.apc.org

