










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Links relacionados
Compartir

 Permalink
PermalinkArchivos de Pediatría del Uruguay
versión impresa ISSN 0004-0584versión On-line ISSN 1688-1249
Arch. Pediatr. Urug. vol.88 no.1 Montevideo feb. 2017
Síndrome de Bartsocas-Papas y secuencia de bridas amnióticas. Semejanzas clínicas sugieren un factor etiopatogénico común
Bartsocas-Papas syndrome and amniotic bands sequence.
Clinical similarities suggest a common etiopathogenic factor
Gabriel Ercoli1, Nancy Mazzitelli2, Mónica Rittler3
1. Médico. Especialista Genética Médica. Centro Nacional de Genética Médica. Buenos Aires. Argentina.
2. Médico. Anatomopatólogo. Hospital Materno Infantil Ramón Sardá. Facultad de Medicina. Universidad de Buenos Aires. Argentina.
3. Médico. Genetista. Hospital Materno Infantil Ramón Sardá. Facultad de Medicina. Universidad de Buenos Aires. Argentina.
Centro Nacional de Genética Médica. Hospital Materno Infantil Ramón Sardá. Facultad de Medicina. Universidad de Buenos Aires. Argentina.
Trabajo inédito.
Declaramos no tener conflictos de interés.
Fecha recibido: 12 de octubre de 2016
Fecha aprobado: 1° de diciembre de 2016
Resumen
Introducción: el síndrome de Bartsocas-Papas (SBP) es una entidad letal causada por una mutación homocigota del gen RIPK4, el cual, formando parte de una compleja red molecular, está involucrado en la diferenciación del queratinocito. Las principales manifestaciones del síndrome, parcialmente compartidas por otras displasias ectodérmicas monogénicas son: pterigium en miembros inferiores, fisuras faciales, defectos de reducción distal de miembros y apéndices cutáneos. Por otro lado, la secuencia de bridas amnióticas (SBA), de etiopatogenia desconocida, se caracteriza por un espectro variable de anomalías consideradas disruptivas, incluyendo fisuras faciales y defectos de reducción de miembros.
Objetivo: describir los hallazgos clínicos y anatomopatológicos de un paciente con SBP y de otro con SBA, remarcando sus semejanzas.
Casos clínicos: 1) Recién nacida con fisura facial, pterigium poplíteo y crural, reducción de dedos y ortejos, y apéndices cutáneos. Placenta con zonas desprovistas de epitelio amniótico. 2) Feto femenino de 27 semanas, fisura facial atípica, reducción distal de miembros, pterigium poplíteo, defecto de cierre de pared tóracoabdominal y polo cefálico, apéndices cutáneos y presencia de bridas amnióticas. Placenta y cordón umbilical con extensas zonas de desprendimiento del epitelio amniótico.
Conclusiones: algunos hallazgos similares en el SBP y
Palabras clave: SÍNDROME DE BARTSOCAS-PAPAS
SÍNDROME DE BANDAS AMNIÓTICAS
Summary
Introduction: Bartsocas-Papas syndrome (BPS) is a lethal condition caused by a homozygous mutation of the RIPK4 gene, which, being part of a complex molecular network, is involved in keratinocyte differentiation while. The main clinical manifestations are webbing of lower limbs, facial clefts, distal limb reduction defects, and skin tags, and some of these are shared with other monogenic ectodermal dysplasia syndromes. Similarly, amniotic bands sequence (ABS), a condition of unknown etiopathogenesis, is characterized by a variable spectrum of anomalies considered as disruptive, such as facial clefts and limb reduction defects.
Objective: to describe clinical and autopsy findings of a patient with BPS and of a fetus with ABS, emphasizing on their similarities. Case reports:
1. Female liveborn with atypical facial clefts, popliteal and crural webbing, distal reduction defects of hands and feet, and multiple skin tags. The placenta showed regions lacking amniotic epithelium. 2. Female fetus of 27 gestational weeks, atypical facial clefts, distal limb reduction defects, popliteal webbing, thoracoabdominal and cephalic closure defects, skin tags, and amniotic bands. Placenta and umbilical cord showed broad regions lacking amniotic epithelium attachment.
Conclusions: the observation of a number of findings with remarkable similarities between BPS and ABS allows considering the existence of an intrinsic factor, involved in skin development which is common to both conditions.
Key words: BARTSOCAS-PAPAS SYNDROME
AMNIOTIC BAND SYNDROME
Introducción
El síndrome de Bartsocas-Papas (SBP), también llamado pterigium poplíteo letal, es una entidad de etiología genética y herencia autosómica recesiva(1,2). Los hallazgos típicos detectables al examen físico son: fisura orofacial, defectos de reducción de manos y pies, pterigium poplíteo y crural, y anomalías cutáneas (alopecia parcial, apéndices cutáneos, bandas filiformes y quistes de milium constituidos por depósitos de queratina y queratinocitos degradados). A pesar de la elevada letalidad neonatal, se han reportado casos con sobrevida de meses y hasta años(3,4). La causa más frecuente del SBP es una mutación en homocigosis del gen RIPK4, localizado en el cromosoma 21q22.3, y que interviene en la diferenciación de los queratinocitos(5,6).
La secuencia de bridas amnióticas (SBA) constituye un conjunto de anomalías fetales consideradas disruptivas, que incluyen fisuras orofaciales, defectos digitales, de cierre del polo cefálico y de la pared tóraco-abdominal(2). Las alteraciones varían en gravedad, originando un espectro.
Se han propuesto diversas hipótesis para explicar su origen(7-9) y, aunque ninguna haya sido universalmente aceptada y que la etiopatogenia de
Objetivos
Describir a una recién nacida con SBP y a un feto con SBA y destacar las semejanzas entre las anomalías congénitas observadas en ambos cuadros. Se especula sobre un posible factor intrínseco común.
Casos clínicos
Caso 1
Recién nacida de término, con peso adecuado para la edad gestacional. Al nacimiento presentó dificultad respiratoria y cianosis, falleciendo a los pocos minutos.
Al examen físico se constató: fisura facial medial, abarcando la región de boca y nariz con ausencia de ambas estructuras (figura 1a); fusión de párpados con probable microftalmia bilateral, un puente cutáneo entre regiones supra e infraoculares derechas, orejas pequeñas e hiperplegadas, pelo escaso y ralo en cuero cabelludo, ausencia de cejas y pestañas, milium generalizado. Defectos distales de los cuatro miembros, con ausencia de dedos y ortejos, manos con lesión cicatrizal en el extremo del muñón y ausencia completa del pulgar. Pterigium poplíteo y crural extensos (figura 2a y b) y múltiples apéndices cutáneos: 1) en puente, uniendo ambos muñones de los pies entre sí y a éstos con la región perineal (figura 3a); 2) pediculado en orificio anal (figura 4a y b); 3) sésiles en la región medial de mentón y tórax (figura 4c).
Radiológicamente, en manos se observó ausencia de todos los componentes del primer rayo digital y de las falanges distales de los 4 dedos restantes; en pies (difícil identificación), ausencia de varios metatarsianos y falanges. Se realizó el diagnóstico de síndrome de Bartsocas-Papas.
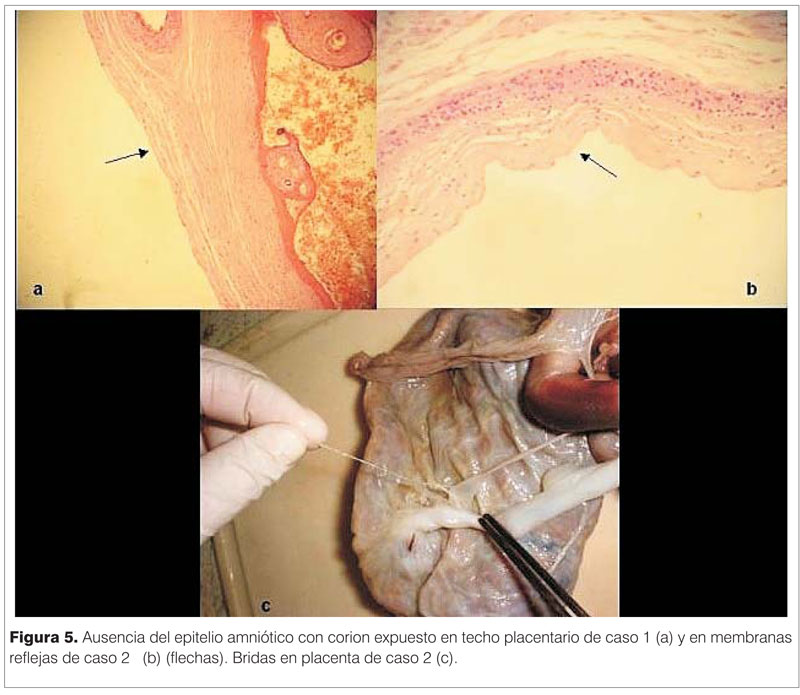
La autopsia no reveló malformaciones internas. Los pulmones eran pequeños, sin cumplir con los criterios estrictos de hipoplasia. Pudo observarse una ligera reducción del calibre laríngeo, en coincidencia con la sospecha clínica de estenosis de la vía respiratoria. La piel (pared abdominal) presentaba una dermis fibrosa y ausencia de anexos en algunas zonas. La histología de los apéndices estaba constituida por una epidermis desde atrófica hasta hiperplásica y un eje conectivo vascular con anexos cutáneos de estructura hamartomatosa. El epitelio amniótico mostró diferentes patrones regionales: en el techo placentario se observaron áreas con ausencia de epitelio (figura 5a); las membranas reflejas presentaban zonas conservadas, otras con hiperplasia del epitelio, con necrosis y con epitelio ausente. La superficie del cordón umbilical se encontraba denudada, con ausencia total del epitelio amniótico, dejando expuesto un tejido conectivo denso e hipercelular subyacente.
Caso 2
Feto femenino de 27 semanas de gestación y peso de
Discusión
Bartsocas-Papas y otros síndromes con displasia ectodérmica
En el ratón, el gen RIPK4 se expresa en la mayoría de los tejidos pero predomina en las capas basales y suprabasal de la epidermis. Su mutación en homocigosis da lugar a fusión digital, así como a atresia de esófago y orificios corporales; la muerte ocurre por asfixia. Existe desorganización de los estratos celulares de la piel y la diferenciación de los queratinocitos es anómala(15).
Por las semejanzas con el SBP, Kalay y colaboradores(5) consideraron al gen RIPK4 humano como candidato etiológico más probable. Mediante la secuenciación de sus 8 exones en individuos afectados, se detectó una transversión T>A en el exón 2, determinando la sustitución de asparagina por isoleucina en el dominio serina/treonina kinasa de la proteína. En otros afectados se identificaron mutaciones de los exones 3 y 5(3), todos correspondientes al mismo dominio de la proteína. La complejidad de la vía de desarrollo de la epidermis resulta evidente por la participación de múltiples moléculas, cuyas mutaciones dan lugar a diversos síndromes con semejanzas clínicas(16). Entre ellas, p63 es un gen regulador maestro, esencial para el desarrollo del epitelio estratificado. Sus mutaciones, las que a su vez afectan la expresión de otros genes, dan lugar a una serie de síndromes caracterizados principalmente por displasia ectodérmica, anomalías digitales y fisuras orales, tales como los síndromes de Hay Wells (anquilobléfaron, displasia ectodérmica, fisura oral)(6,17,18) y EEC (ectrodactilia, displasia ectodérmica, fisura oral)(2); p63 a su vez activa a RIPK4, a NF-KB (factor nuclear kappa B) y a IRF6 (factor regulador de interferón 6). La interacción de este último con RIPK4 sería un requisito para prevenir la fusión epitelial característica en los síndromes con pterigium(19) y sus mutaciones dan lugar a los síndromes de Van der Woude (fisura labiopalatina y fístulas del labio inferior(15,20,21) y Escobar (pterigium poplíteo no letal)(2). NF-KB, en cuya activación también interviene RIPK4 y cuyas mutaciones muestran semejanzas clínicas con el SBP, es un regulador de diversos procesos, entre los cuales se cuentan la proliferación y apoptosis en varios tejidos, la diferenciación de queratinocitos y la formación de la cresta ectodérmica apical, estructura fundamental para el desarrollo de los miembros(5,22); p63 controlaría además la expresión de genes desmosomales, función que podría explicar la fragilidad cutánea y la desorganización de las capas epidérmicas en los síndromes con displasia ectodérmica(23).
Secuencia de bandas amnióticas
Los cuadros clínicos diagnosticados como SBA abarcan un amplio espectro, desde una simple reducción digital hasta múltiples defectos de miembros, de pared tóraco-abdominal y polo cefálico y fisuras orofaciales. Con frecuencia se asocian malformaciones de diversos órganos internos(2). Resulta necesario aclarar que el diagnóstico de SBA es particularmente arbitrario, ya que las bridas amnióticas, capaces de reforzarlo, no siempre están presentes. Se han mencionado tres teorías que intentan explicar su patogenia: 1) Teoría intrínseca o de displasia del disco germinal(7). El origen sería un defecto intrínseco del embrión, con expresión tanto embrionaria como extraembrionaria. Las alteraciones involucrarían a la piel fetal y a las membranas amnióticas, así como al desarrollo de otros órganos y estructuras. Aunque el hipotético factor intrínseco no ha sido identificado, se trataría de un componente genético y no puede descartarse su intervención en la cascada molecular responsable del desarrollo epidérmico. 2) Teoría extrínseca o de disrupción mecánica(8): Todos los defectos se deberían a una ruptura prematura del amnios, su separación del corion y la formación de bandas fibrosas, causantes de amputaciones y constricciones. Ciertas partes fetales se introducirían en el espacio entre amnios y corion y la estrecha aposición de partes fetales al corion generaría abrasiones cutáneas, favoreciendo la adhesión de las bandas amnióticas a las regiones denudadas. Las anomalías observadas dependerían de la edad gestacional al momento de la ruptura del amnios. Los defectos craneofaciales y de la pared abdominal serían de origen temprano, mientras que la ruptura tardía afectaría solamente a los miembros.
Esta teoría, sin embargo, no logra explicar el saco amniótico intacto observado en algunos casos, ni las frecuentes malformaciones de órganos internos. 3) Teoría vascular(9): el origen de
Semejanzas clínicas entre síndromes con displasia ectodérmica y
Según algunos autores,
Comparación de los aspectos genéticos
El SBP y otros síndromes con displasia ectodérmica son de etiología monogénica, significando que la sola mutación del gen en cuestión es responsable del fenotipo. La recurrencia familiar es frecuente, dependiendo de la forma de herencia determinada por el tipo de mutación. Contrariamente, la recurrencia familiar de
Proponemos que
Conclusiones
En base a las semejanzas observadas entre las displasias ectodérmicas monogénicas y
Agradecimientos
A las doctoras Carmen Gutiérrez por sus sugerencias acerca de la redacción del manuscrito y Liliana Vauthay por sus aportes a la discusión sobre los mecanismos del desarrollo embrionario.
Referencias bibliográficas
1. Bartsocas C, Papas C. Popliteal pterygium syndrome. Evidence for a severe autosomal recessive form. J Med Genet 1972; 9(2):222-6.
2. Johns Hopkins University. OMIM Online Mendelian Inheritance in Man: an online catalog of human genes and genetic disorders. Baltimore, MD: Johns Hopkins Medicine, 2015. Disponible en: https://www.omim.org/. (Consulta: 20 diciembre 2015).
3. Abdalla E, Morsy H. Bartsocas-Papas syndrome: unusual findings in the first reported egyptian family. Case Rep Genet 2011;2011:428714.
4. Giannotti A, Digilio MC, Standoli L, Zama M, Dallapiccola B. New case of Bartsocas-Papas syndrome surviving at 20 months. Am J Med Genet 1992; 42(5):733-5.
5. Kalay E, Sezgin O, Chellappa V, Mutlu M, Morsy H, Kayserili H, et al. Mutations in RIPK4 cause the autosomal-recessive form of popliteal pterygium syndrome. Am J Hum Genet 2012; 90(1):76-85.
6. Mitchell K, O’Sullivan J, Missero C, Blair E, Richardson R, Anderson B, et al. Exome sequence identifies RIPK4 as the Bartsocas-Papas syndrome locus. Am J Hum Genet 2012; 90(1):69-75.
7. Streeter G. Focal deficiencies in fetal tissues and their relation to intra-uterine amputation. Contrib Embryol 1930; 22(1):1-44.
8. Torpin R. Amniochorionic mesoblastic fibrous strings and amnionic bands: associated constricting fetal malformations or fetal death. Am J Obstet Gynecol 1965; 91:65-75.
9. Van Allen MI, Curry C, Gallagher L. Limb body wall complex: I. Pathogenesis. Am J Med Genet 1987; 28(3):529-48.
10. Etches P, Stewart A, Ives E. Familial congenital amputations. J Pediatr 1982; 101(3):448-9.
11. Lubinsky M, Sujansky E, Sanger W, Salyards P, Severn C. Familial amniotic bands. Am J Med Genet 1983; 14(1):81-7.
12. Irving W, Doublestein G. Congenital amniotic band syndrome: report of a familial recurrence. J Am Osteopath Assoc 1988; 88(7):891-3.
13. Levy R, Lacombe D, Rougier Y, Camus E. Limb body wall complex and amniotic band sequence in sibs. Am J Med Genet A 2007; 143A(22):2682-7.
14. Blyth M, Lachlan K. Amniotic bands in paternal half-siblings. Clin Dysmorphol 2010; 19(2):62-4.
15. Rountree R, Willis C, Dinh H, Blumberg H, Bailey K, Dean C Jr, et al. RIP4 regulates epidermal differentiation and cutaneous inflammation. J Invest Dermatol 2010; 130(1):102-12.
16. Rinne T, Hamel B, van Bokhoven H, Brunner H. Pattern of p63 mutations and their phenotypes—update. Am J Med Genet A 2006; 140(13):1396-406.
17. McGrath JA, Duijf P, Doetsch V, Irvine A, de Waal R, Vanmolkot K, et al. Hay-Wells syndrome is caused by heterozygous missense mutations in the SAM domain of p63. Hum Mol Genet 2001; 10(3):221-9.
18. Moretti F, Marinari B, Lo Iacono N, Botti E, Giunta A, Spallone G, et al. A regulatory feedback loop involving p63 and IRF6 links the pathogenesis of 2 genetically different human ectodermal dysplasias. J Clin Invest 2010; 120(5):1570-7.
19. De Groote P, Tran H, Fransen M, Tanghe G, Urwyler C, De Craene B, et al. A novel RIPK4-IRF6 connection is required to prevent epithelial fusions characteristic for popliteal pterygium syndromes. Cell Death Differ 2015; 22(6):1012-24.
20. Richardson R, Dixon J, Malhotra S, Hardman MJ, Knowles L, Boot-Handford R, et al. Irf6 is a key determinant of the keratinocyte proliferation-differentiation switch. Nat Genet 2006; 38(11):1329-34.
21. Biggs L, Rhea L, Schutte B, Dunnwald M. Interferon regulatory factor 6 is necessary, but not sufficient, for keratinocyte differentiation. J Invest Dermatol 2012; 132(1):50-8.
22. Bushdid P, Brantley D, Yull F, Blaeuer G, Hoffman L, Niswander L, et al. Inhibition of NF-kappaB activity results in disruption of the apical ectodermal ridge and aberrant limb morphogenesis. Nature 1998; 392(6676):615-8.
23. Ferone G, Mollo MR, Thomason H, Antonini D, Zhou H, Ambrosio R, et al. p63 control of desmosome gene expression and adhesion is compromised in AEC syndrome. Hum Mol Genet 2013; 22(3):531-43.
24. Bamforth JS. Amniotic band sequence: Streeter’s hypothesis reexamined. Am J Med Genet 1992; 44(3):280-7.
25. Luehr B, Lipsett J, Quinlivan JA. Limb-body wall complex: a case series. J Matern Fetal Neonatal Med 2002; 12(2):132-7.
Correspondencia: Dr. Gabriel Ercoli.
Correo electrónico: gabrielercoli312@hotmail.com


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}