











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo

 Permalink
Permalink
Introducción
El hemo es un compuesto químico muy importante que forma parte de múltiples hemoproteínas, como hemoglobina, mioglobina, citocromos y catalasas. Se sintetiza en el hígado y en la médula ósea e involucra ocho enzimas, cuatro mitocondriales y cuatro citosólicas1.
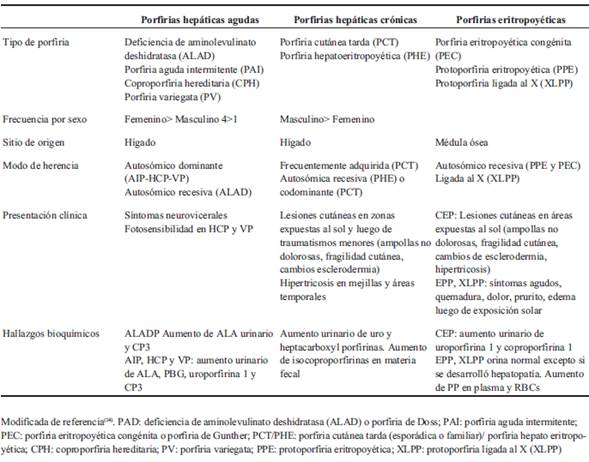
Las porfirias son un grupo de enfermedades hereditarias o adquiridas, ocasionadas por alteraciones en la actividad de las enzimas que participan en la vía de síntesis del hemo (Figura 1). Existen siete tipos de porfirias con gran heterogeneidad clínica. Se clasifican según su forma de presentación clínica o según el órgano mayormente afectado2) (Tabla 1). La forma más frecuente es la porfiria cutánea tarda (PCT).

La porfiria hepato eritropoyética (PHE) es una variante de la PCT3,4. Su mecanismo de herencia es autosómico y recesivo. Se trata de un tipo muy raro de porfiria, con poco más de 40 casos reportados en el mundo. Las características clínicas más frecuentes son el inicio precoz en la infancia -antes de los 2 años-, hipertricosis, lesiones cutáneas tipo ampollas, erosiones en áreas expuestas al sol o de traumatismos con cicatrización atrófica tipo quiste de milium y compromiso hepático3. La PHE se debe a un déficit en la actividad de la enzima uroporfirinógeno decarboxilasa (UROD), quinta enzima en el proceso de síntesis del hemo (Figura 1). La actividad enzimática es muy baja, por lo que su presentación clínica es más precoz que la PCT5.
El diagnóstico de la PHE requiere en primer lugar de un alto grado de sospecha clínica y luego de la realización de análisis bioquímicos, enzimáticos y moleculares1. El tratamiento es sintomático, la fotoprotección de la piel es una medida esencial, acompañada de tratamientos médicos, entre ellos la hidroxicloroquina1.
Se presenta un caso de una PHE en la edad pediátrica. El objetivo es describir una enfermedad muy rara, las principales características clínicas que deben orientar el diagnóstico para iniciar un tratamiento precoz y realizar asesoramiento genético. En nuestro conocimiento, se trata del primer caso de PHE reportado en nuestro país.
Caso clínico
Paciente de 5 años, sexo femenino, procedente de Montevideo. Sin antecedentes perinatales ni personales a destacar, niega ingesta de fármacos. Adecuado crecimiento y desarrollo psicomotor. Carné de inmunizaciones vigente. Padres consanguíneos. Antecedentes por línea materna de una tía y bisabuela con enfermedad hepática de etiología desconocida. Inicia manifestaciones clínicas a los 3 años, caracterizadas por hipertricosis en áreas expuestas al sol, principalmente en mejillas, área fronto-temporal, antebrazos y tronco (Figura 2). Agrega en la evolución lesiones ampollares en dorso de manos, no dolorosas, con cicatrización posterior tipo quistes de milium (Figura 3). No presenta eritrodoncia ni orinas rojizas. Valorada en varias oportunidades, se realiza diagnóstico de impétigo y otras infecciones de piel. Recibe múltiples planes antibióticos, sin que se obtenga respuesta, por lo que es derivada a dermatología. Se plantea sospecha de porfiria de manifestación cutánea. Se realizan serologías para descartar factores precipitantes de porfiria: serología para VIH, hepatitis B y C, que son negativas.


Se solicitan estudios más específicos. Porfirinas en orina: uroporfirinas 113 (g/24 h (rango normal de 0-40 (g/24 h) y coproporfirinas 2 (g/24 h (rango normal de 0-160 (g/24 h). Dado el cuadro clínico y el aumento de uroporfirinas en orina, se solicita estudio genético, que reporta variante patogénica en homocigosis para gen UROD: c.185 C>T, p. (Pro62Leu). Entre otros estudios realizados se destaca: hemograma, función renal, funcional y enzimograma hepático, metabolismo de hierro y glicemia, normales. Ecografía abdominal sin alteraciones. En la evolución instala diabetes insulino dependiente estabilizada con insulina cristalina y NPH, en seguimiento con endocrinólogo. A los 4 años se inicia tratamiento con hidroxicloroquina con mejoría progresiva de las lesiones de piel e hipertricosis (Figura 4) y (Figura 5). Se realizan controles regulares con funcional y enzimograma hepático. Presenta hepatitis como efecto adverso, por lo que se suspende el fármaco. Se reinicia luego en forma gradual asociando S-acetil-metionina (SAMe), sin cambios posteriores en el funcional hepático y buena evolución de la enfermedad. El asesoramiento genético es de alto riesgo de recurrencia para PHE para la pareja.


Discusión
Las porfirias son un grupo complejo y clínicamente heterogéneo de enfermedades que se caracterizan por defectos en la síntesis del grupo hemo por deficiencia de alguna de las enzimas que intervienen en el proceso. El lugar donde falle la vía metabólica determinará el tipo de metabolito intermediario que se acumule y, consecuentemente, la presentación clínica (Figura 1). Dada la baja incidencia y la variabilidad de sintomatología es necesario un alto índice de sospecha clínica para establecer el diagnóstico2.
Dentro de este grupo de enfermedades solo algunas se presentan en la edad pediátrica: PEC, PPE, XLPP y PHE6.
La prevalencia global de las porfirias se desconoce, pero se estima entre 1:500 a 1:50.000 según la población y el tipo de porfiria7. La PCT es la más frecuente de todos los subtipos y puede ser congénita o adquirida, con esta última forma como la más común. El consumo de alcohol, tabaco, estrógenos, presentar infecciones como hepatitis C, entre otros, se describen como factores predisponentes para las formas adquiridas, descritas en adultos8,9.
Las porfirias pueden ser hepáticas o eritroides, según el órgano mayormente afectado. En las formas hepáticas, aunque el defecto se encuentra en el hígado, también pueden presentar manifestaciones cutáneas. En las formas eritroides, además de la afectación cutánea, puede existir compromiso medular con anemia que llegue a requerir transfusiones frecuentes2.
La presentación clínica con lesiones tipo ampollas y erosiones es característico de las porfirias cutáneas. Las lesiones son indoloras, con piel frágil, se localizan especialmente en dorso de mano, dejan cicatrices hipo o hiperpigmentadas tipo quistes de milium. La fragilidad cutánea con lesiones cicatrizales puede en ocasiones confundirse con maltrato (lesiones por quemadura de cigarrillo). La hipertricosis se localiza principalmente en rostro, cola de ceja, mejillas y en casos típicos se dispone como un antifaz6. Ambas son de importante valor semiológico en el diagnóstico de la enfermedad4,10. El mecanismo patogénico de las manifestaciones de piel en las porfirias cutáneas está relacionado con el efecto de la luz solar sobre el exceso de porfirias en las células de la piel11. Justamente, las lesiones de piel y la hipertricosis fueron los elementos clínicos que en nuestra paciente llevaron al planteo de porfiria y a la orientación de los estudios más específicos. Hay casos reportados en los que el diagnóstico se confirma incluso varios años después del inicio de los síntomas12,13. En el caso que presentamos hubo aproximadamente un año de retraso en el diagnóstico.
En la PHE puede existir hepatitis al momento del diagnóstico; en este caso el funcional y enzimograma hepático fueron inicialmente normales. Se destaca que, a diferencia de la PCT, en la PHE, no hay sobrecarga férrica3.
Los estudios específicos para el diagnóstico de la enfermedad son la cuantificación de porfirinas en orina y materia fecal (Tabla 1). En este caso el hallazgo de uroporfirinas elevado en orina orientó hacia porfirias cutáneas de origen hepático: PCT o PHE.
Existen otros análisis, como la cuantificación de heptacarboxyl porfirinas en orina y de isocoproporfirinas en materia fecal, que permiten apoyar el diagnóstico si están aumentados4. La confirmación definitiva se realiza mediante la secuenciación del gen UROD ubicado en 1p34.1 que codifica para la enzima del mismo nombre. En la PHE la enzima UROD tiene una actividad de alrededor del 5%, a diferencia de la PCT, con una actividad enzimática en torno al 50%. Como en otras enfermedades metabólicas, dicha diferencia en la actividad enzimática puede explicar el inicio clínico más precoz en la infancia para la PHE14.
En la paciente descrita se identificó una variante en homocigosis para este gen. Esta ha sido reportada en homocigosis en dos medios hermanos de una familia portuguesa con PHE, y se clasifica como variante patogénica PMID: (8644733)15. Confirmado el diagnóstico, se realiza asesoramiento genético a la familia. De la historia familiar de la paciente, destacamos que se trata de una pareja consanguínea, elemento que debe hacer sospechar de una enfermedad genética recesiva. Por otro lado, hay antecedentes de enfermedad hepática crónica de etiología no aclarada por línea materna. Por ello, en este contexto, consideramos de relevancia continuar el abordaje diagnóstico de esta familia.
En cuanto al tratamiento de la PHE, se recomienda como primera medida la fotoprotección. Otro tratamiento utilizado ha sido la hidroxicloroquina a dosis bajas, que actúa movilizando las porfirias hepáticas del compartimento intracelular, lo que facilita su excreción urinaria1,3,16. Se describen situaciones en las que el uso de hidroxicloroquina no ha sido beneficioso y se ha observado elevación transitoria inicial de transaminasas hepáticas17. En el caso de la paciente se iniciaron las medidas de fotoprotección así como hidroxicloroquina. Se evidenció una rápida mejoría clínica, como puede verse en las Figuras 3 y 4. La adición de SAMe concomitantemente con la reintroducción de la hidroxicloroquina luego del aumento de transaminasas permitió la estabilidad y tolerancia posterior. El SAMe es un compuesto que disminuye el estrés oxidativo a nivel hepático18. Su uso ha sido reportado en algunos casos de PHE en la edad pediátrica19,20.
No hay consenso sobre la duración del tratamiento con hidroxicloroquina. Singal AK y col. compararon diferentes tratamientos para la PCT y encontraron que con la administración de hidroxicloroquina se objetiva la remisión clínica de los síntomas en aproximadamente seis meses, con un descenso de las porfirinas urinarias de forma concomitante8.
Se ha descrito que la diabetes mellitus tipo 2 puede estar presente hasta en el 25% de los pacientes con PCT, pero no se ha reportado relación con la PHE21. La paciente que reportamos tuvo inicio de diabetes mellitus tipo I que desconocemos si podría estar relacionado con la PHE.
En cuanto al seguimiento, se recomiendan controles periódicos con equipo interdisciplinario: monitorear la funcionalidad hepática, las complicaciones endocrinológicas así como controles oftalmológicos periódicos, dado que el efecto adverso más importante del uso crónico de hidroxicloroquina es el daño ocular con compromiso de retina22-24.
Conclusión
Presentamos el caso clínico de una niña con una enfermedad de base genética muy rara, como es la PHE. Presenta manifestaciones cutáneas que es necesario conocer para realizar diagnósticos diferenciales con enfermedades pediátricas frecuentes.
Se trata de una afección compleja que requiere un abordaje interdisciplinario con dermatólogos, endocrinólogos, pediatras, genetistas, especialistas en errores innatos del metabolismo y apoyo psicológico.
Por el momento no tiene un tratamiento curativo y puede asociar importantes secuelas y complicaciones. El abordaje del núcleo familiar es de fundamental importancia así como acompañar el proceso de la enfermedad.

