











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo

 Permalink
Permalink
Importancia del tema
Las alteraciones de la forma del cráneo son un motivo frecuente de consulta, y suponen un reto para el clínico que las enfrenta. Es fundamental conocer los estudios iniciales que se deben realizar y cual es el momento oportuno para derivar a los distintos especialistas.
El clínico debe saber diferenciar cuando una alteración en la forma del cráneo se debe a una causa intrínseca secundaria a una fusión prematura de una o más suturas, a cuando dicha alteración se debe a una causa extrínseca que ocasiona una deformidad posicional1.
En nuestro medio, no se cuenta actualmente con protocolos de manejo de alteraciones de la forma del cráneo. Por eso es de real importancia la realización de una guía clínica para el manejo.
Los objetivos de esta guía son:
- Identificar a los recién nacidos y lactantes con alteraciones de la forma del cráneo.
- Analizar los estudios paraclínicos disponibles en nuestro medio para la valoración de dichos pacientes.
- Proponer un algoritmo de manejo y derivación oportuna en recién nacidos con alteraciones de la forma del cráneo.
Introducción
Normalmente el desarrollo craneal comienza entre la tercera y cuarta semana de gestación con la formación de siete huesos, que van a constituir lo que conocemos como bóveda craneal (dos huesos parietales, dos temporales, dos frontales y un occipital).
Existen cuatro suturas mayores (metópica, sagital, coronal y lambdoidea) y tres suturas secundarias (frontonasal, escamosa, esfenofrontal). La sutura metópica separa ambos huesos frontales, la sagital separa ambos huesos parietales, la coronal separa el hueso frontal del parietal y la sutura lambdoidea el parietal del occipital. Además de los huesos y suturas se encuentran las fontanelas, destacando la anterior o bregmática (delimitada por huesos frontales y parietales) y la posterior o lambdoidea (delimitada por los huesos parietales y occipitales)2. Esto se muestra en la (Figura 1).

En condiciones fisiológicas las suturas y fontanelas craneales progresan hacia el cierre, con períodos de fusión independientes para cada una de ellas. La sutura metópica es la primera en cerrarse, entre los 3 y 8 meses de vida. Luego comienzan a cerrarse la sutura sagital a los 22 meses, la coronal a los 24 meses y la sutura lambdoidea a los 26 meses aproximadamente. El cierre de las suturas secundarias comienza en la esfenofrontal a los 22 meses, en la sutura escamosa entre los 35-39 meses y en la frontonasal a los 68 meses aproximadamente. Según la clasificación de Meindl, Lovejoy y White, el cierre de las suturas continúa hasta los 40 años2,3.
Definimos craneosinostosis como la fusión prematura, intrauterina, de una o más suturas del cráneo. La sinostosis determinará que el cráneo no crezca de forma perpendicular a la sutura involucrada, con un aumento compensatorio de las suturas abiertas, lo que se conoce como la ley de Virchow. Esto no siempre se acompaña de menor volumen encefálico o crecimiento craneal1,4.
La craneosinostosis es la segunda anomalía craneofacial más común, después de las hendiduras orofaciales, con una prevalencia de 1 en 2.250 nacimientos vivos. Ocurre en todos los grupos étnicos5.
Si bien la mayoría de las complicaciones de las craneosinostosis son estéticas y funcionales, también pueden generar alteraciones motoras, del lenguaje o neurocognitivas, sobre todo vinculadas a las que afectan varias suturas. Esto último también ocurre en las sinostosis que comprometen una única sutura, principalmente en relación al cierre de la sutura coronal o lambdoidea5-7.
Existen diversas formas de clasificar a las craneosinostosis, todas complementarias. Se las puede dividir en primaria o secundaria, simple o compleja, y sindrómica o no sindrómica4,8.
La craneosinostosis primaria ocurre por alteraciones genéticas, que actúan solas o en combinación con factores ambientales. La secundaria se debe a trastornos que afectan la sutura en desarrollo, como trastornos hematológicos (talasemia, enfermedad de células falciformes), metabólicos (hiperparatiroidismo, hipercalcemia, deficiencia de vitamina D, hipofosfatasia, pseudohipoparatiroidismo), displasias óseas (acondroplasia, mucopolisacaridosis, mucolipidosis) y exposición a fármacos (fenitoína, valproico, retinoides, etcétera)6,9-12.
La sinostosis simple es cuando se afecta una sola sutura, mientras que en las complejas se afecta más de una.
Las sinostosis sindrómicas implican la fusión de una o múltiples suturas, sumada a la presencia de otras anomalías (malformaciones en los miembros, cardíacas, del SNC y traqueales). Este grupo de craneosinostosis están más relacionadas con aumento de la presión intracraneal, cefaleas, ceguera, alteraciones en el neurodesarrollo, pérdida auditiva, y afectación de la vía respiratoria alta (secundaria a la hipoplasia mediofacial y anomalías de la tráquea), que puede conducir a apneas, asfixia e incluso la muerte5,7,13,14.
Algunos trabajos han planteado que se subestima la incidencia de la hipertensión endocraneana que generan las sinostosis, pudiendo generar consecuencias para el desarrollo normal del cerebro, principalmente secuelas neurocognitivas15,16.
Evaluación clínica
Es importante realizar una correcta anamnesis para seguir con el examen físico.
Hay ciertas preguntas que pueden ayudar a diferenciar una craneosinostosis de una deformidad posicional2,4.
1) ¿La alteración del cráneo está presente desde el nacimiento?
2) ¿Hay una preferencia en la posición al dormir?
3) ¿Hay una mejoría con el paso del tiempo?
La craneosinostosis está presente desde el nacimiento y empeora a medida que transcurre el tiempo.
Es importante en esta etapa reconocer factores de riesgo como alteraciones del útero materno, posición al nacer, restricciones intrauterinas (gestación múltiple, oligohidramnios, movimientos fetales), duración de la gestación, tamaño fetal, afectación en familiares, enfermedad tiroidea, tabaquismo, exposición a teratógenos (warfarina, nitrofurantoína, ácido valproico, etcétera)1,17,18.
Se reconocen como factores de riesgo para la plagiocefalia posicional el sexo masculino, la prematurez, la tortícolis y la hipotonía10.
Es importante reconocer los elementos de alarma que requieren intervenciones de forma aguda, como la disminución del nivel de conciencia, déficit neurológico, dificultad respiratoria, asfixia, vómitos e irritabilidad, que pueden asociarse frecuentemente a sinostosis sindrómicas10.
En cuanto al examen físico, en primer lugar se debe realizar la inspección del cráneo desde todas las posiciones (adelante, atrás, lateral y especialmente desde arriba, que es donde se aprecian mejor las alteraciones). Se valorará la forma de la cabeza, el tamaño y la simetría de las órbitas oculares, la simetría de la base del cráneo y la posición de las orejas. Hay que palpar las suturas y fontanelas para corroborar si existe un engrosamiento o crestas por fusión prematura1.
El análisis de fotografías puede aportar datos sobre si la asimetría estaba presente desde el nacimiento y cómo ha sido su evolución.
La medición del perímetro cefálico ha mostrado baja confiabilidad, alta variabilidad interobservador y baja sensibilidad para detectar craneosinostosis19.
El craneómetro se suele utilizar para la medición craneal luego de confirmarse una plagiocefalia posicional no sinostósica para establecer el grado de deformidad (Figura 2). En la plagiocefalia posterior se medirá la diferencia de diámetros oblicuos, siendo leve si es menor de 10 mm, moderada entre 10 a 20 mm y grave si es mayor a 20 mm. En la braquicefalia posicional, el índice craneal se calcula con el cociente entre ancho craneal y la longitud multiplicado por 100, siendo leve entre 80-89, moderado entre 90-100 y grave mayor a 100. Esta clasificación es importante para establecer el tratamiento más adecuado en función de la gravedad1.

Es importante evaluar los rasgos faciales minuciosamente, así como buscar malformaciones que involucren a la vía respiratoria, auditiva, extremidades y otros sistemas (cardiovascular, genitourinario, digestivo)10.
Diagnósticos diferenciales
Las fuerzas mecánicas que se ejercen durante el trabajo de parto y parto, principalmente de tracción y compresión, pueden causar diversas patologías que conllevan alteraciones de la forma del cráneo.
Algunos factores de riesgo son la macrosomía fetal, el trabajo de parto prolongado, el parto instrumental (utilización de fórceps) y la presentación podálica.
Las más frecuentes son la bolsa serohemática, el cefalohematoma y la remodelación craneal con cabalgamiento de suturas.
La bolsa serohemática es una colección serosanguinolenta subcutánea y extraperióstica (Figura 3). Presenta una distribución con bordes mal definidos, sin respetar suturas. La piel puede presentar equimosis o hematomas en la zona afectada. Rara vez presentan complicaciones en la evolución, se resuelven de forma espontánea en pocos días y sin tratamiento20.

El cefalohematoma es una hemorragia traumática subperióstica, que afecta más frecuentemente al hueso parietal (Figura 3). Generalmente es unilateral y respeta suturas. Puede estar asociado a una fractura lineal del cráneo. Su resolución en la mayoría de los casos es espontánea, pudiendo durar hasta 6 meses20.
La remodelación craneal con cabalgamiento de suturas aparece como consecuencia de los cambios transitorios del cráneo que sufre el recién nacido al momento del parto. Hay un aumento de la presión anteroposterior y en menor medida transversal21.
Otros diagnósticos diferenciales menos frecuentes son la microcefalia, la macrocefalia, la hidrocefalia y el encefalocele.
Etiología
Se han propuesto múltiples hipótesis para explicar la patogénesis de la fusión prematura de las suturas. Hoy en día la craneosinostosis se entiende como un trastorno oligogénico, multifactorial y heterogéneo. Factores genéticos y epigenéticos, como fuerzas mecánicas e interacciones ambientales externas, participan en su desarrollo (Tabla 1). También se propone el concepto de “matriz funcional”, donde fuerzas morfológicas y funcionales se combinan para producir el resultado anatómico final. El avance genético ha permitido reconocer múltiples genes involucrados en el desarrollo óseo y de las suturas. Aun así en el 70% de los pacientes afectados se desconoce la etiología(2, 12,17).

La formación de las suturas craneales requiere de moléculas osteoinhibidoras y osteoinductoras que regulan el crecimiento y la formación de los huesos del cráneo (factor de unión a la heparina soluble, FGF2, y TGF-b), de vías de señalización que participan en la diferenciación ósea (TGF-b/BMP, Wnt, Hedgehog, Notch y FGFs), así como de genes (TWIST1) involucrados en la migración temprana y supervivencia de las células madre mesenquimales12.
La craneosinostosis no sindrómica es la más frecuente, explica aproximadamente el 90% de los casos, mayormente con afectación de una única sutura. Aproximadamente menos de 10% corresponde a causas familiares, mientras que el resto ocurre de forma esporádica. Los determinantes genéticos y ambientales contribuyen al fenotipo. Se han descrito distintas mutaciones monogénicas y alteraciones cromosómicas, aunque la mayoría de los determinantes genéticos aún son desconocidos. En los últimos años se reconoció al gen TCF12 como responsable importante de la sinostosis coronal no sindrómica1,12,13,17.
La mayoría de las craneosinostosis sindrómicas son secundarias a alteraciones monogénicas autosómicas dominantes, con penetrancia incompleta y expresividad variable; aunque también se han reconocido mutaciones de novo y alteraciones cromosómicas. Múltiples genes, como FGFR1, FGFR2, FGFR3, TWIST1, MSX2 y EFNB1, se han involucrado en este tipo de craneosinostosis. Por otro lado, en un tercio de las sinostosis de la sutura coronal se han detectado mutaciones monogénicas. La sinostosis coronal es el tipo más comúnmente observado en los síndromes de Saethre-Chotzen, Muenke y los craneofrontonasales. Las alteraciones cromosómicas, representan entre 6,7%-40% y se presentan principalmente con sinostosis de las suturas metópicas o sagitales. La alteración cromosómica más frecuente es la delección o translocación de la región 7p21 que incluye gen TWIST111,12,17.
Craneosinostosis no sindrómicas
Se abordarán las sinostosis de las grandes suturas, dejando de lado las suturas secundarias ya que son muy poco frecuentes y generalmente no tienen un fenotipo característico dado que cuando se ven afectadas suelen estar comprometidas otras suturas22.
Sinostosis sagital
Es la más común de las sinostosis no sindrómicas, dando cuenta de un 50-60% del total de las mismas. Se presenta en 1 cada 2.000 nacimientos, siendo el sexo masculino el más afectado. La sinostosis de esta sutura da como resultado un cráneo estrecho en sentido bitemporal y alargado en sentido anteroposterior, adoptando forma de “bote o casco de un navío”, denominado escafocefalia o dolicocefalia (Figura 4)1,2,5.

Sinostosis coronal
Predomina en el sexo femenino. La forma que adopta el cráneo varía si la afectación es uni o bilateral. La afectación unilateral se denomina plagiocefalia anterior, y es la tercera sinostosis más frecuente. En estos casos el cráneo toma un aspecto oblicuo en su sector anterior, provocado por un aplanamiento ipsilateral de la frente, con elevación del ala esfenoidal y del techo de la órbita del mismo lado, dando el aspecto de que la órbita está más elevada. También la raíz nasal se desvía hacia el lado de la sutura cerrada y hay una protuberancia compensatoria de la frente contralateral (Figura 4)1,2.
En los casos de afectación bilateral, el cráneo adopta una forma conocida como braquicefalia, produciéndose un acortamiento anteroposterior del cráneo secundario al aplanamiento a nivel frontal. Se observa una frente plana, con elevación de las cejas y protrusión orbitaria, pudiendo presentar aspecto turricefálico cuando aumenta la altura de la cabeza de forma compensatoria (Figura 4)1,2.
Sinostosis metópica
Ha ido en aumento en los últimos años y es la segunda más frecuente, con una incidencia de 1 cada 5.200 nacimientos. Es más frecuente en sexo masculino. Su fusión precoz produce un cráneo de forma triangular o trigonocefalia, en el que se evidencia una cresta en la línea media de la frente, un estrechamiento frontotemporal, una constricción del pterion, hipotelorismo y aumento del diámetro biparietal (Figura 4)1,2.
Ocasionalmente se puede desarrollar una cresta metópica visible o palpable en la región frontal, permaneciendo la morfología craneal normal. En esta variante de la normalidad, la sutura puede parecer fusionada en las pruebas de imagen, pero algunos estudios histopatológicos han demostrado que no existe una sinostosis verdadera1.
Sinostosis lambdoidea
Puede existir afectación uni o bilateral. La sinostosis lambdoidea unilateral o plagiocefalia posterior, es la menos frecuente, presentando una incidencia de 1 en 40.000 nacimientos (Figura 4) y (Figura 6).
En estos casos hay un aplanamiento del occipucio homolateral, abombamiento de la apófisis mastoides del mismo lado e inclinación de la base del cráneo hacia abajo en el lado alterado con desplazamiento posteroinferior de la oreja ipsilateral y prominencia frontal contralateral. La sinostosis lambdoidea bilateral es rara y provoca una región occipital aplanada con ensanchamiento occipitoparietal y elongación del vertex. Esto conlleva mayor riesgo de malformación de Chiari tipo I y fusión del agujero yugular, lo que genera mayor probabilidad de aumento de la presión intracraneana1,2.
Craneosinostosis sindrómicas
Hay más de 150 síndromes craneofaciales conocidos asociados a craneosinostosis, dentro de los más frecuentes se incluyen el síndrome de Apert, Crouzon, Pfeiffer, Muenke y Saethre-Chotzen (Tabla 2)23.
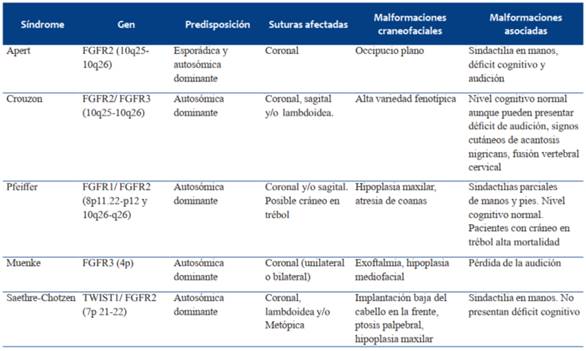
Síndrome de Apert
Puede afectar a 1 de cada 65.000 nacimientos. Se asocia a edad paterna avanzada.
La mayoría de las mutaciones ocurren de forma esporádica, aunque también se puede heredar de forma autosómica dominante. Se han detectado 4 mutaciones del gen FGFR2 que causan este síndrome, localizadas en 10q25-10q26. Las características clínicas del cráneo dependen de las suturas que están involucradas, pero la mayoría de los casos se presentan al nacimiento con braquiturricefalia y occipucio plano, dando lugar a una frente alta y grande, exoftalmia y estrabismo divergente. Asocian hipoplasia mediofacial, con una boca de forma trapezoidal. La sindactilia simétrica bilateral de manos es característica de este síndrome. Presentan habitualmente déficit cognitivo y disminución de la audición24,25.
Síndrome de Crouzon
Se debe a mutaciones en el gen FGFR2, que se heredan de forma autosómica dominante. Presenta una amplia variedad fenotípica. Con frecuencia compromete el desarrollo craneofacial. En comparación con el síndrome de Apert, habitualmente se trata de una sinostosis compleja, con afectación de múltiples suturas, dando lugar a un volumen craneal menor. A nivel cervical puede presentarse fusión vertebral. Estos pacientes suelen tener un desarrollo cognitivo normal, aunque generalmente presentan una audición disminuida. Una forma menos frecuente del síndrome de Crouzon se presenta con signos cutáneos de acantosis pigmentaria que aparece durante los primeros 2 años de vida y se deben a una mutación en FGFR324.
Síndrome de Pfeiffer
Es causado por mutaciones en los genes FGFR1 y FGFR2, localizados en el cromosoma 8p11.22-p12 y 10q26-q26 respectivamente, las cuales se heredan de forma autosómica dominante. Generalmente se presenta con afectación de la sutura coronal, con o sin afectación de la sagital.
Cohen ha descrito tres grupos clínicos, con distintas implicancias pronósticas. El tipo 1 tiene el fenotipo clásico, con braquiturricefalia, hipoplasia maxilar, atresia de coanas y sindactilias parciales de manos y pies e inteligencia normal. Este subtipo es compatible con la vida. El tipo 2 se asocia a cráneo en hoja de trébol y afectación del SNC. Los niños afectados en general mueren de forma precoz. El tipo 3 es similar al último, pero sin cráneo en hoja de trébol. Los subtipos 2 y 3 son extremadamente raros24.
Síndrome de Muenke
Es el síndrome más frecuente de todos, afectando a 1 de cada 30.000 nacimientos. Es ocasionado por una sola mutación del gen FGFR3 localizado en el cromosoma 4p, que se hereda de forma autosómica dominante. Al igual que el síndrome de Apert, se asocia a edad paterna avanzada. Hay afectación de la sutura coronal de forma uni o bilateral, con grados variables de exoftalmia, sin hipoplasia mediofacial apreciable. Es frecuente la pérdida de la audición por afectación neurosensorial24.
Síndrome de Saethre-Chotzen
Es causado por una mutación de herencia autosómica dominante a nivel del gen TWIST 1 en el cromosoma 7p 21-22. Se presenta con craneosinostosis de la coronal, lambdoidea o metópica. Suelen presentar implantación baja del cabello en la frente, ptosis palpebral, hipoplasia maxilar con paladar estrecho, anomalías de la columna cervical y malformaciones digitales, como sindactilia del segundo y tercer dedo de la mano, así como duplicación del primer dedo del pie. Pueden presentar alteraciones en el desarrollo y en el aprendizaje, pero el deterioro cognitivo no es típico24.
Plagiocefalias posicionales no sinostósicas
La deformidad posicional del cráneo es la principal causa de asimetría. Es ocasionada por causas extrínsecas que presionan sobre los huesos blandos y maleables de la calota del lactante en los primeros meses de vida. Tiene una incidencia de 1 en 300 nacidos vivos, y ha aumentado considerablemente en los últimos años debido a las pautas actuales, que apuntan a disminuir el riesgo de muerte súbita del lactante. En consecuencia, si bien se ha reducido drásticamente el riesgo de muerte súbita, la incidencia de plagiocefalia posicional ha aumentado en un 600%1,25.
Los factores de riesgo pueden ser prenatales, o más comúnmente posnatales. Dentro de los primeros se destacan las anomalías uterinas, embarazo gemelar, oligoamnios, presentación podálica e hipotonía.
Algunos de estos factores de riesgo pueden provocar alteraciones en la forma del cráneo evidentes desde el nacimiento. Los factores posnatales están principalmente vinculados con el tiempo en posición decúbito supino, asimetría en la lateralización cefálica, retraso motor y apoyo cefálico sobre superficies duras haciendo que se presente la deformidad craneal en las siguientes semanas o meses al nacimiento1.
Es importante aclarar que el término plagiocefalia hace referencia a una asimetría en la forma del cráneo, pudiendo ser predominantemente anterior o posterior. Dicha asimetría puede ser secundaria a una plagiocefalia posicional, así como a una sinostosis coronal o lambdoidea unilateral6,26.
La plagiocefalia posicional posterior (PPP) es la alteración más frecuente del cráneo. En este caso la alteración del cráneo, con aplanamiento unilateral parietooccipital, se suele acompañar de desplazamiento anterior de la oreja y prominencia frontal del mismo lado, lo que determina una forma del cráneo romboidal o de paralelogramo (Figura 5) y (Figura 6). Ante la preferencia de la posición cefálica se debe descartar una tortícolis asociada1.


Figura 6: Diferencias semiológicas entre la plagiocefalia posicional posterior y sinostosis lambdoidea unilateral.
La braquicefalia posterior (BP) se caracteriza por un aplanamiento simétrico de toda la zona occipital, con un occipucio como “cortado por un hacha” (Figura 5). Presenta además un ensanchamiento bilateral en las regiones témporoparietales, generando un cráneo corto y ancho. Con frecuencia se puede observar un abultamiento por encima de las orejas1.
La braquicefalia asimétrica es una combinación de la plagiocefalia y la braquicefalia, caracterizada por un aplanamiento occipital asimétrico acompañado de ensanchamiento parietal bilateral (Figura 5)27.
La escafocefalia o dolicocefalia deformativa se observa con más frecuencia en prematuros con internaciones prolongadas en unidades neonatales, con compresiones biparietales que dan lugar al aspecto de cráneo alargado por aumento del diámetro anteroposterior y disminución del biparietal (Figura 5)1.
Métodos diagnósticos
La radiografía de cráneo fue clásicamente el método de estudio inicial, pero hoy no se utiliza, dados los nuevos métodos más precisos. Puede ser útil para el diagnóstico de sinostosis sagitales, coronales o lambdoideas. No es de gran utilidad para el diagnóstico de sinostosis metópica, debido al cierre temprano de esta sutura. En los casos raros o de difícil diagnóstico, la realización de radiografías de buena calidad con cuatro enfoques (anteroposterior, Towne y dos proyecciones laterales), puede ser suficiente para excluir craneosinostosis y evitar más radiaciones. Si el estudio no es concluyente por la corta edad del paciente, se recomienda repetir las radiografías luego de 1 o 2 meses1,2,4,7,28.
Las impresiones digitales en radiografías no son patognomónicas de HTE, y se pueden observar en niños sanos. Lo mismo que las imágenes en TC que dependen de la ventana ósea29,30.
La tomografía con reconstrucción tridimensional (TC 3D) es el método con mayor sensibilidad y especificidad. Permite visualizar todas las suturas, examinar el sistema ventricular, la posición del tronco encefálico y cerebelo, así como anomalías cerebrales de la línea media. No se recomienda como método de screening. Sus principales indicaciones son ante casos en los que la clínica no es concluyente, así como para valoración prequirúrgica y postoperatoria. Nuevas técnicas de reconstrucción que utilizan bajas dosis de irradiación, han logrado disminuir la exposición a la misma, sin comprometer la calidad de imagen1,2,10,23,26,31-34.
La ecografía con enfoque de suturas es un método rápido, inocuo y de bajo costo, que permite evaluar estructuras superficiales. Cuando es realizada por personal con experiencia muestra una elevada sensibilidad y especificidad, ambas por encima del 90%, para confirmar sinostosis. La sutura normal se ve como una brecha o espacio hipoecoico entre dos placas óseas hiperecoicas. Una sutura sinostósica muestra la pérdida de dicho espacio entre las placas óseas. También puede observarse una cresta ósea. Debe ser considerado como el estudio de imagen inicial en pacientes menores de 12 meses con sospecha de craneosinostosis. Hoy en día no se recomienda como estudio de tamizaje prenatal dada la baja sensibilidad de la técnica. Los síndromes asociados a craneosinostosis son raros y difíciles de diagnosticar antes del tercer trimestre. En estos pacientes se puede utilizar la ecografía y la resonancia prenatal en forma complementaria1,18,28,32,35-37.
La resonancia magnética de cráneo es el estudio de elección para valorar el parénquima encefálico, pero no es el ideal para la evaluación de sinostosis, y además requiere sedación del paciente. Está indicada cuando se evidencian alteraciones neurológicas o del desarrollo que sugieren una posible afectación del sistema nervioso central asociada. Recientemente se introdujo la secuencia “BlackBox”, que minimiza el contraste de los tejidos blandos para mejorar la imagen de los bordes óseos, planteándose como posible sustituto de la tomografía, así también como una base para modelos impresos en 3D8,31,38-40.
La estereofotogrametría o fotografía 3D es una técnica no invasiva, utilizada para evaluar y medir el cráneo. La técnica implica el uso de dos cámaras digitales, que toman imágenes de la cabeza del paciente desde diferentes ángulos. Luego las imágenes en 3D se analizan en una computadora. Estudios recientes postulan a este método como posible sustituto de la tomografía19,38.
El volumen intracraneal se ha utilizado para evaluar el crecimiento craneal, pero requiere de imágenes de resonancia magnética o tomografía para su cálculo. Además existen estudios que demuestran que los niños con craneosinostosis tienen con frecuencia un volumen intracraneano en rangos normales19.
Los estudios genéticos pueden aportar datos importantes en el diagnóstico etiológico. Se deben considerar ciertos factores como las suturas involucradas, anomalías asociadas, hitos del desarrollo, así como la historia familiar, para seleccionar los pacientes candidatos a realizar test genéticos18,41.
Los estudios de laboratorio como hormonas tiroideas, calcio, fosfato, fosfatasa alcalina, niveles de vitamina D son de utilidad si existe sospecha por el interrogatorio y examen físico10.
Tratamiento
La Academia Americana de Pediatría (AAP) ha establecido guías para el manejo de las plagiocefalias posicionales no sinostósicas. En los casos leves a moderados se recomienda terapia conservadora con medidas de reposicionamiento y fisioterapia. Esta última es igual de efectiva que el reposicionamiento con almohadas, pero cumple además con las recomendaciones de sueño seguro de la AAP. La deformidad mejorará a medida que avancen los hitos motores gruesos del desarrollo1,42.
Se debe insistir en la educación a los padres y prevención primaria desde las unidades neonatales. Se recomienda alternar la posición para dormir, colocando al niño en decúbito supino en sueño, girando la cabeza a ambos lados y estimular el decúbito prono en vigilia. Si el lactante presenta tortícolis se recomienda la fisioterapia para mejorar el rango de movimiento del cuello1.
En los casos de deformidad moderada a grave sin mejoría con tratamiento conservador, se recomienda la órtesis craneal (OC) mediante el uso de casco. La misma ha demostrado beneficio y bajas complicaciones en el tratamiento, sin embargo su indicación debe ajustarse a la relación costo-beneficio. El casco se debe usar 23 horas al día, adaptándose a la medida de la cabeza del niño, con comprensión en las áreas sobreexpandidas y mayor espacio para las zonas planas. Se recomienda su uso desde los 4 meses de vida, y por un máximo de 12 meses1,42-44.
En los casos de craneosinostosis confirmada, se decidirá en conjunto con equipo craneofacial la indicación, oportunidad y técnica quirúrgica. La familia también tomará parte de esta decisión. La clave para poder ofrecer todas las opciones terapéuticas es el diagnóstico precoz, así como la derivación oportuna1.
La mayoría de los autores considera que la cirugía craneofacial es una cirugía funcional. La indicación de este tratamiento tiene como objetivo corregir de forma estética y reparadora la deformidad craneal. En ocasiones también se busca evitar o minimizar los impactos negativos del aumento de la presión intracraneana, así como del desarrollo neurocognitivo y del deterioro visual1,45-47.
La oportunidad quirúrgica dependerá del tipo y gravedad de la sinostosis, así como de las técnicas utilizadas por el cirujano. La cirugía temprana permite actuar cuando la deformidad craneal es menos grave y aún no se ha desarrollado incremento de la PIC, dado que el riesgo de desarrollar hipertensión endocraneana es menor en los primeros seis meses1,48.
Numerosos estudios han demostrado un mejor resultado estético y funcional si la cirugía es antes del primer año de vida, debido a que el cerebro cuadriplica su volumen en los primeros tres años49.
La craneotomía abierta con reconstrucción, así como los procedimientos endoscópicos mínimamente invasivos, son las opciones terapéuticas. Existe heterogeneidad de técnicas. Antes de los 5-6 meses de vida se pueden realizar técnicas mínimamente invasivas, ya que los huesos craneales son aún lo suficientemente flexibles y pueden ser manipulados por el endoscopio. Este tipo de técnicas tienen a favor una reducción del tiempo operatorio, menor pérdida de sangre, alta hospitalaria más temprana, así como resultados cosméticos y funcionales excelentes. Estos procedimientos requieren un dispositivo secundario para alterar la forma del cráneo con el tiempo, que incluye cascos ortopédicos, resortes y brazos de distracción osteogénica46,47,50,51.
La reconstrucción de la bóveda craneal implica la eliminación de la sutura fusionada y el hueso craneal deformado, seguido de la remodelación con reemplazo óseo. Este tipo de intervenciones quirúrgicas se realizan típicamente entre los 6 y 12 meses de vida, ya que se necesita que el hueso sea más sólido, permitiendo una reconstrucción con menores riesgos y recidivas. Luego de los 12 meses los procedimientos realizados tienen mayor riesgo de defectos craneales persistentes1,46,48,51.
En las craneosinostosis sindrómicas generalmente son necesarias múltiples cirugías y la oportunidad operatoria puede ser incluso tan precoz como en el período neonatal. Los enfoques terapéuticos suelen variar en diferentes centros, ya que algunos están a favor del abordaje temprano para favorecer el mejor crecimiento cerebral, mientras que otros ponderan el abordaje tardío con mejores resultados de reconstrucción ósea. Esto suele ser controvertido debido a que no existen estudios prospectivos comparativos53,54.
Dentro de las posibles complicaciones del tratamiento quirúrgico se encuentran: infecciones de la herida, sangrados, hematomas (subgaleal, subdural), hipertermia, rotura de duramadre, pérdida de líquido cefalorraquídeo y meningitis51.
La mortalidad posoperatoria oscila en 2,6%, mientras que la morbilidad posoperatoria en un 12%51.
Son pacientes complejos que requieren siempre de un equipo multi e interdisciplinario: pediatra, neonatólogo, neuropediatra, neurocirujano, anestesista pediátrico, cirujano plástico, otorrinolaringólogo, oftalmólogo, hemoterapeuta, fisioterapeuta, asistente social, psicología médica, entre otros.
Se ha visto que la consulta tardía con equipo especializado contribuye notoriamente a peores resultados. En Uruguay se observó que la edad promedio de consulta fue de 15 meses, y muchos de estos niños se intervinieron quirúrgicamente alrededor de los dos años de vida (datos sin publicar).
En la década de 1980, en el ámbito de la UDELAR-Hospital de Clínicas, se creó el Centro de Estudio y Tratamiento de Afecciones Craneofaciales (CETAC), en una gran mancomunión de todas las especialidades nombradas anteriormente. A la fecha ha realizado cerca de 600 cirugías, con resultados comparables a los centros internacionales y con una morbimortalidad similar (datos sin publicar).
Luego de la cirugía es necesario el seguimiento de los pacientes de forma regular, controlando el crecimiento y el perímetro craneano, observando posibles signos de aumento de la presión intracraneana, así como otras potenciales complicaciones. La reosificación incompleta es otro elemento por el cual se deben controlar estos pacientes a largo plazo (Tabla 3)51-54.

Por último, proponemos el algoritmo de evaluación y derivación oportuna en pacientes con alteración de la forma del cráneo que se muestra en la (Figura 7).


