











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo

 Permalink
Permalink
Introducción
La enfermedad de Von Willebrand adquirido (EVW adquirida) es un trastorno hemorrágico poco frecuente adquirido, informado por primera vez en 1968, con características clínicas y de laboratorio similares a la Enfermedad de Von Willebrand hereditaria 1,2).
La EVW adquirida es causada por una deficiencia del factor de von Willebrand (FVW) cuantitativa o cualitativa. Los factores principales que distinguen esta enfermedad incluyen la falta de trastornos hemorrágicos previos, el diagnóstico a una edad avanzada, no contar con antecedentes familiares y la asociación con condiciones subyacentes.
El diagnóstico debe sospecharse en adultos con sangrado mucocutáneo inusual asociado a trastornos linfoproliferativos, mieloproliferativos, autoinmunes y cardiovasculares 3. Los trastornos linfoproliferativos y mieloproliferativos representan alrededor del 50-60% de todos los casos reportados 4).
El Mieloma múltiple (MM) es una neoplasia clonal de células plasmáticas en la médula ósea, caracterizada típicamente por inmunoglobulinas séricas anormales en sangre periférica o parte de ésta. Es una neoplasia maligna tratable, pero incurable.
El MM es la única discrasia de células plasmáticas clonales que siempre está precedido por una fase premaligna, denominada gammapatía monoclonal de significado incierto (MGUS), pero no siempre detectable. MGUS está presente en más del 3% de la población por encima de los 50 años y progresa a MM o enfermedad maligna relacionada en una tasa de 1% por año. Dado que MGUS es asintomático, más del 50% de los individuos al diagnóstico con MGUS tenían esta condición durante más de 10 años antes del diagnóstico clínico. La supervivencia del MM mejoró con la introducción de fármacos inmunomoduladores e inhibidores del proteosoma en la década anterior, pero la tasa actual de supervivencia a 5 años no supera el 50% 5,6).
Se expondrá a continuación un caso clínico que involucra estas enfermedades.
Caso clínico
Paciente de sexo femenino, de 57 años, con antecedentes personales de gastritis diagnosticada por endoscopía digestiva hace dos años en tratamiento con omeprazol. Ex tabaquista. Colecistectomizada. Anemia leve crónica tratada en alguna oportunidad con hierro. Sin antecedentes gineco-obstétricos relevantes, sin historia de sangrados patológicos.
Ingresó por un síndrome hemorragíparo dado por equimosis fáciles de reciente aparición y por sangrado prolongado de una semana de evolución posterior a exodoncia de pieza dental del maxilar inferior que no cede con medidas locales.
Presentaba fatiga moderada sin otros elementos de síndrome funcional anémico.
Del examen físico se destacaba palidez cutáneo mucosa, hematomas aislados de pequeño tamaño evolucionados en miembros superiores e inferiores. Sangrado activo a nivel gingival. Sin repercusión hemodinámica.
De la paraclínica al debut destacaba una anemia crónica agudizada dado por una caída de 2 puntos del valor basal alcanzado un valor de hemoglobina de 6 g/dl, pura normocítica y normocrómica, plaquetas 172.000/mm3, leucocitos 6,700/mm3. Crasis: TTPA 46 segundos, TP 95,7%, INR 1,02, Fibrinógeno 306. Debido al TTPA prolongado, se realizó Test de mezcla con plasma normal, donde se observó corrección del parámetro, correspondiendo a un déficit de los factores de la coagulación.
La paciente ingresó para valoración diagnóstica. La actividad coagulante del factor VIII (FVIII: C) fue de 23%, el antígeno del factor von Willebrand (vWF: Ag) 12 % y la actividad del cofactor de Ristocetina fue de 11%.
Se confirmó la coagulopatía, haciendo diagnóstico de Enfermedad de Von Willebrand adquirida a una enfermedad desconocida.
En el funcional y enzimograma hepático se observó hiperproteinemia, proteínas totales de 11g/dl. Ante la sospecha de EVW adq. por una patología hemato-oncológica, se solicitó proteinograma electroforético que evidenció hiperproteinemia total con una banda de aspecto monoclonal de 5.90 g/dL en la zona de migración de las beta 2 globulinas e hipogammaglobulinemia. Inmunoglobulinas séricas con elevación de la concentración de IgM 9870 mg/dl, IgA 32 mg/dl e IgG 266 mg/dl. Cadenas livianas libres en suero Kappa 16,48 mg/L, lambda 4,75 mg/L, ratio Kappa/Lambda 3,47. Ionograma y función renal normal. Serologías virales negativas.
Se realizó interconsulta al equipo de Hematología ante el planteo de que ésta coagulopatía sea secundaría a una gammapatía monoclonal.
Dada la necesidad de realizar un procedimiento invasivo solicitado para valoración diagnostica, Biopsia de médula ósea (BMO), en contexto de un pico monoclonal IgM elevado se realizaron dos recambios plasmáticos terapéuticos (RPT) con la extracción de una volemia plasmática por procedimiento. Se obtuvo como resultado una disminución en la concentración del pico monoclonal (de 5.97 a 2.93 mg/dl) y aumento en el nivel de los factores de la coagulación FVIII: C 54%, FVW: Ag 18%, FVW: CoR 21%. Debido al antecedente de sangrado gingival grave con requerimiento transfusional, se administró una dosis de 500UI/1200UI de FVIII/FVW como tratamiento profiláctico inmediatamente previo a la biopsia junto con ácido tranexámico. No presentando complicaciones hemorrágicas durante ni luego del procedimiento.
De los estudios realizados se destacaba: Mielograma/BMO: infiltración de casi el 100% de células plasmáticas. Los estudios de Inmunotipificación, Inmunofenotipo e Inmunohistoquimica, confirmaron la clonalidad de las células y alejó diferenciales a otras neoplasias de células B.
Se confirmó el diagnóstico de EVW adquirida Secundaria a Mieloma múltiple IgM Estadio de Durie Salmon III-A ISS I, IgM Kappa (CM: 5,9 g/dl).
Para la estratificación pronostica se obtuvieron resultados con diferimiento en el tiempo de: LDH 213 U/L, beta 2 microglobulina 2,7 mg/L, albúmina 4,10 g/dl, estudio citogenético sin alteraciones y FISH no evaluable. Proteínas en orina de 24 horas (PU24) 0,04 g/L, 0,14 g/24hs no significativa.
TC de bajas dosis: múltiples lesiones líticas en cráneo, cervicales, lumbo-sacra, pelvis no afectación de muro posterior, ni cortical.
De acuerdo a la clasificación de la IMWG se confirmó el diagnóstico de un MM activo, la anemia de etiología multifactorial podría considerarse como un criterio CRAB, además de considerar los marcadores biológicos infiltración medular de más del 60%, ratio FLC 3.47.
Para recordar la estratificación R-ISS aún usada en la actualidad, corresponde a un estadío I esta paciente (supervivencia global -OS- a 5 años 82%, supervivencia libre de progresión -SLP- a 5 años 55%); véase como en este particular es adecuado el uso de ISS (que prescinde del resultado citogenético) estadio I, (mediana de supervivencia 62 meses)
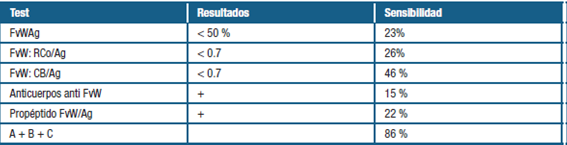
Se adjunta imagen ilustrativa de un modelo vigente para comprender los mecanismos subyacentes a la EVW adquirida (tabla 1 y 2)
Tabla 1: Causas y mecanismos fisiopatológicos de Enfermedad Von Willebrand adquirida. Extraído de: Moro et al. Enfermedad de Von Willebrand adquirida en un linfoma linfoplasmocitario/Macroglobulinemia de Waldenström: reporte de caso. 7
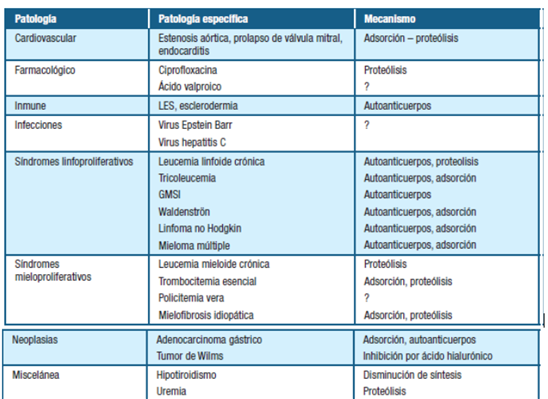
Tabla 2: Sensibilidad de las pruebas de laboratorio en Enfermedad Von Willebrand adquirida. Extraído de: Moro et al. Enfermedad de Von Willebrand adquirida en un linfoma linfoplasmocitario/Macroglobulinemia de Waldenström: reporte de caso. (7)
La paciente fue valorada de forma multidisciplinaria guiado por Hemoterapia y Hematología. El tratamiento inicialmente estuvo destinado a la corrección de la coagulopatía, para detener el sangrado agudo y comenzar un tratamiento precoz de la patología de base. Recibió 1 unidad de sangre desplasmatizada (SD) y plasma fresco congelado (PFC) 10 ml/kg, lo que optimizó el manejo de la anemia y la corrección parcial de la crasis.
La paciente recibió RPT que se utilizó para disminuir los niveles elevados de la paraproteína IgM, evitar un síndrome de hiperviscosidad, y porque debía realizarse un procedimiento invasivo como la BMO. El procedimiento de RPT se realizó con un separador celular Spectra Optia ®, y fue una herramienta terapéutica eficaz para reducir la concentración de ésta inmunoglobulina de distribución principalmente intravascular (en un 80%) 8,9,10.
Se realizaron dos recambios plasmático terapéuticos, de una volemia por procedimiento, como la paciente no presentaba sangrado activo hace varias semanas se utilizó como solución de reemplazo albúmina al 4%, sin complicaciones hemorrágicas. Se obtuvo como resultado una disminución en la concentración del pico monoclonal y un aumento de los factores de la coagulación.
El perfil de la EVW adquirida corresponde a un tipo 1, nivel del FVW < 0.30 Ul/ml, relación CO:R/Ag:FVW > 0,7. En pacientes sometidos a cirugía menor o procedimientos invasivos menores, se considera aumentar los niveles de actividad del FVW > 0.50 UI/mL con desmopresina o concentrado de factor con la adición de ácido tranexámico 11).
En este caso se utilizó concentrado de factor VIII/FVW, (Haemate P®, CSL BEHRING S.A.) dada la severidad de sangrado presentado, administrando una única dosis de 500 Ul/1200 UI de FVIII/FVW previo a la BMO. Haemate-P es un concentrado de FVIII/FVW, deshidratado, pasteurizado, estéril y liofilizado, derivado del plasma humano, que se utiliza administrado por vía intravenosa en el tratamiento de pacientes con hemofilia A o EVW. Como agente coadyuvante se empleó ácido tranexámico, 500 mg vía oral cada 8 horas desde las 24 hs previas a la BMO y c/8 hs por 5 días.
En cuanto al tratamiento de la patología subyacente la paciente comenzó con un plan de poliquimioterapia en base a Ciclofosfamida 500 mg vía oral, Bortezomib 2,3 mg subcutáneo y Dexametasona 40 mg vía oral. Se realizaron las solicitudes correspondientes al Fondo Nacional de Recursos para lograr optimizar el tratamiento de inducción con la adición de un inmunomodulador e inhibidor de protesoma.
Discusión
La EVW adquirida al igual que la forma congénita, cursa con un defecto cuantitativo o cualitativo del FVW, una gran glicoproteína adhesiva que interviene en la hemostasia primaria y secundaria, actuando en la adhesión de las plaquetas al vaso sanguíneo, y es chaperona del FVIII de la coagulación respectivamente.
Existen tres grandes categorías de EVW según la deficiencia del FVW, tipo 1 el más frecuente (déficit cuantitativo parcial), tipo 2 (déficit cualitativo), y el tipo 3 (déficit cuantitativo total). En el caso descrito, se trataría de una EVW adquirida de tipo 1. Según reportes internacionales el patrón más frecuentemente descrito es de una EVW adquirida de tipo 2. Se desconoce la verdadera prevalencia de la EVW adquirida, ya que muchos casos pueden ser silenciosos desde el punto de vista clínico y permanecer sin diagnosticar 3).
El diagnóstico debe sospecharse en adultos con sangrado mucocutáneo inusual asociado a trastornos linfoproliferativos, mieloproliferativos, autoinmunes y cardiovasculares.
Los mecanismos de disminución del FVW pueden ser de etiología inmune o no inmune, y muchas veces coexisten. En las enfermedades neoplásicas el mecanismo más probable propuesto es la presencia de Autoanticuerpos contra el FVW pero estos sólo se pueden confirmar en 20% de los casos debido a técnicas de baja sensibilidad y especificidad 12. Estos son mayormente de tipo IgG, raramente IgM, y excepcionalmente IgA. Otra causa involucrada en linfomas es la adsorción del FvW por células malignas. Mediante la expresión de moléculas de adhesión aberrantes en la superficie de las células tumorales se produce la unión del FvW a las mismas, lo que determina su disminución en plasma 7).
En nuestro caso no se demostró un inhibidor y APTT con pool de plasma normal supone un mecanismo de adsorción del FVW por las células tumorales.
Dentro de las neoplasias malignas hemato-oncológicas con un pico IgM inicialmente, por frecuencia, las neoplasias linfoides fueron el primer planteo. La Macroglobulinemia de Waldenström (MW)/ Linfoma linfoplasmocítico, Linfoma de la zona marginal. Más alejados estadios pre malignos: gammapatía monoclonal de significado incierto (MGUS) o mieloma latente (SMM) pero se alejaban por la presencia de anemia crónica si era considerada como elemento de daño de órgano blanco.
El estudio citomorfologico así como la ausencia de la mutación MYD88L265P apoyó el diagnóstico del debut de MM IgM. Se remarca que la mutación MYD88L265P no es patognomónica de las discrasias plasmáticas ni de linfomas indolentes, se observa en el linfoma esplénico de la zona marginal (4%), amiloidosis IgM (71%), linfoma de tejido linfático asociado a la mucosa (7%) y MW (67% -90%). Se puede concluir que la mutación MYD88 L265P está asociada específicamente con MW e IgM MGUS y su ausencia apoya al planteo de ésta excepcional entidad MM IgM. Es útil para orientar a próximos estudios y encaminar una estrategia costo-efectiva.
Las pistas útiles para el diagnóstico de MM incluyen la presencia de lesiones óseas líticas (raras en MW) y una translocación en t (11; 14) (no ocurre en MW). Los pacientes con MM IgM tienden a tener una diferenciación plasmocítica con alta expresión de CD138 e inmunoglobulina citoplásmica, mientras que MW expresa CD20.
La paciente no contaba con estudios inmunoproteicos previos, ni imagenológicos que hicieran plantear la evolución clonal de un estadio premaligno, destacando además que no todo MGUS progresa a MM, 1% por año puede progresar a MM 13). El Smoldering mieloma SMM (mieloma quiescente-latente) ocupa un lugar intermedio entre MGUS y MM sin signos de daños en órganos diana. El diagnóstico precoz, su estatificación oportuna de acuerdo al riesgo de progresión y el seguimiento estrecho es la conducta actualmente aceptada, si bien hay una tendencia a realizar el tratamiento precoz ya en estos estadios 13).
El tratamiento de EVW adquirida se basa en el tratamiento de la enfermedad de base, pero ante la urgencia como el Síndrome de Hiperviscosidad o ante maniobras invasivas el control de la coagulopatía es prioritario. Además, permite el inicio precoz del tratamiento, en este caso con poliquimioinmunoterapia en la internación.
El MM es una enfermedad incurable y la supervivencia ha mejorado significativamente en los últimos 15 años gracias al desarrollado múltiples combinaciones de inmuno-poliquimioterapia y eventualmente el trasplante de progenitores hematopoyéticos (TPH), que logran la remisión de la enfermedad. Sin embargo, en su mayoría los pacientes recaen, y la duración de la remisión en el MM recidivante disminuye con cada régimen. Por tanto, actualmente su manejo es como una enfermedad crónica.
El conocimiento de las alteraciones genéticas y moleculares abre camino a nuevas terapias blanco “target” farmacológicos y de estratificación para una medicina personalizada. Múltiples alteraciones que involucran la cadena pesada de la inmunoglobulina se han descrito como de mal pronóstico. Se destaca la t (11:14) que además de ser útil en la valoración diagnostica está incluida en scores pronostico como mSMART propuesto por la Clínica Mayo.
El tratamiento del MM ha crecido exponencialmente en los últimos años, reflejándose en la sobrevida de los pacientes, incluso fallecen por otras entidades y no por la progresión del MM. La respuesta es monitorizada a través de los resultados inmunoproteicos, con diferente grado de respuesta. La valoración y utilidad clínica por EMR (enfermedad mínima residual) aún no es de consenso.
Diferentes son los esquemas de tratamiento. Actualmente la clásica división de ser candidato o no a trasplante pauta la inducción, consolidación y mantenimiento. En los países que cuentan con los recursos suficientes, se ha incorporado la droga Daratumumab al clásico triplete de inducción, constituido por un inmunomodulador (ej Lenalidomida), un inhibidor de proteosomas (ej Bortezomib) y dexametasona. Se destaca que existe a nivel internacional planes iniciales de cuatro drogas ejemplo adición de Carfilzomib en paciente de muy alto riesgo, entre otras drogas.
Si bien el TPH es una conducta aceptada de forma precoz (ante 4 ciclos) en algunos casos es válido aplazarlo a 6 ciclos (considerando características del paciente y de la agresividad de la enfermedad). Actualmente podemos generalizar que el gold estándar del régimen de acondicionamiento para un TPH exitoso en esta patología se rige por el aporte de Melfalan, MEL-140 o MEL 200 (mg/m2) de acuerdo a edad, comorbilidad y función renal. En nuestro país el TPH autólogo se realiza en un área de internación especializada, sabiendo que esta terapéutica tiende a tener su manejo en el ámbito ambulatorio a nivel internacional 14).
El mantenimiento con Lenalidomina o Botezomib, en nuestro país es sustentado por el Fondo Nacional de Recursos (FNR), Lenalidomida de administración oral para paciente de bajo riesgo como es ésta paciente mejora la SLP y la OS. Tiene un rol central para mantener la respuesta alcanzada tras el TPH, incluso recomendado para aquellos que no alcanzan una muy buena respuesta parcial -VGPR-. Sin embargo, debe sopesarse su dosificación y frecuencia, porque en múltiples oportunidades las citopenias como toxicidad limitan su administración, además de recordar el aumento de riesgo de cánceres secundarios asociados a la terapia. Aunque es claro el beneficio del mantenimiento faltan datos sobre la duración óptima. Mútliples ensayos también están examinando si la duración del mantenimiento puede modificarse en función de los resultados de la EMR, así como el beneficio de nuevas drogas.
Conclusiones
Se presentó el caso de una paciente sin historia previa de sangrados, que consultó por un síndrome hemorragíparo a la que se le diagnosticó una Enfermedad de von Willebrand adquirido secundario a un Mieloma múltiple IgM kappa en estadío avanzado. Ambas patologías son de muy baja frecuencia, y el abordaje multidisciplinario permitió su estudio integral y el inicio de una terapia adaptada al riesgo.

